Qi Yun, Si-Fei Ma, Wei-Ning Zhang, Meng Gu, Jia Wang
{"title":"作为阿尔茨海默病潜在治疗靶点的 FoxG1:通过AMPK/mTOR自噬途径调节NLRP3炎症体","authors":"Qi Yun, Si-Fei Ma, Wei-Ning Zhang, Meng Gu, Jia Wang","doi":"10.1007/s10571-024-01467-4","DOIUrl":null,"url":null,"abstract":"<p>An increasing body of research suggests that promoting microglial autophagy hinders the neuroinflammation initiated though the NLRP3 inflammasome activation in Alzheimer’s disease (AD). The function of FoxG1, a crucial transcription factor involved in cell survival by regulating mitochondrial function, remains unknown during the AD process and neuroinflammation occurs. In the present study, we firstly found that Aβ peptides induced AD-like neuroinflammation upregulation and downregulated the level of autophagy. Following low-dose Aβ25–35 stimulation, FoxG1 expression and autophagy exhibited a gradual increase. Nevertheless, with high-concentration Aβ25–35 treatment, progressive decrease in FoxG1 expression and autophagy levels as the concentration of Aβ25–35 escalated. In addition, FoxG1 has a positive effect on cell viability and autophagy in the nervous system. In parallel with the Aβ25–35 stimulation, we employed siRNA to decrease the expression of FoxG1 in N2A cells. A substantial reduction in autophagy level (Beclin1, LC3II, SQSTM1/P62) and a notable growth in inflammatory response (NLRP3, TNF-α, and IL-6) were observed. In addition, we found FoxG1 overexpression owned the effect on the activation of AMPK/mTOR autophagy pathway and siRNA-FoxG1 successfully abolished this effect. Lastly, FoxG1 suppressed the NLRP3 inflammasome and enhanced the cognitive function in AD-like mouse model induced by Aβ25–35. Confirmed by cellular and animal experiments, FoxG1 suppressed NLRP3-mediated neuroinflammation, which was strongly linked to autophagy regulated by AMPK/mTOR. Taken together, FoxG1 may be a critical node in the pathologic progression of AD and has the potential to serve as therapeutic target.</p>","PeriodicalId":9742,"journal":{"name":"Cellular and Molecular Neurobiology","volume":"303 1","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-04-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"FoxG1 as a Potential Therapeutic Target for Alzheimer’s Disease: Modulating NLRP3 Inflammasome via AMPK/mTOR Autophagy Pathway\",\"authors\":\"Qi Yun, Si-Fei Ma, Wei-Ning Zhang, Meng Gu, Jia Wang\",\"doi\":\"10.1007/s10571-024-01467-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>An increasing body of research suggests that promoting microglial autophagy hinders the neuroinflammation initiated though the NLRP3 inflammasome activation in Alzheimer’s disease (AD). The function of FoxG1, a crucial transcription factor involved in cell survival by regulating mitochondrial function, remains unknown during the AD process and neuroinflammation occurs. In the present study, we firstly found that Aβ peptides induced AD-like neuroinflammation upregulation and downregulated the level of autophagy. Following low-dose Aβ25–35 stimulation, FoxG1 expression and autophagy exhibited a gradual increase. Nevertheless, with high-concentration Aβ25–35 treatment, progressive decrease in FoxG1 expression and autophagy levels as the concentration of Aβ25–35 escalated. In addition, FoxG1 has a positive effect on cell viability and autophagy in the nervous system. In parallel with the Aβ25–35 stimulation, we employed siRNA to decrease the expression of FoxG1 in N2A cells. A substantial reduction in autophagy level (Beclin1, LC3II, SQSTM1/P62) and a notable growth in inflammatory response (NLRP3, TNF-α, and IL-6) were observed. In addition, we found FoxG1 overexpression owned the effect on the activation of AMPK/mTOR autophagy pathway and siRNA-FoxG1 successfully abolished this effect. Lastly, FoxG1 suppressed the NLRP3 inflammasome and enhanced the cognitive function in AD-like mouse model induced by Aβ25–35. Confirmed by cellular and animal experiments, FoxG1 suppressed NLRP3-mediated neuroinflammation, which was strongly linked to autophagy regulated by AMPK/mTOR. Taken together, FoxG1 may be a critical node in the pathologic progression of AD and has the potential to serve as therapeutic target.</p>\",\"PeriodicalId\":9742,\"journal\":{\"name\":\"Cellular and Molecular Neurobiology\",\"volume\":\"303 1\",\"pages\":\"\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2024-04-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cellular and Molecular Neurobiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10571-024-01467-4\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular and Molecular Neurobiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10571-024-01467-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
FoxG1 as a Potential Therapeutic Target for Alzheimer’s Disease: Modulating NLRP3 Inflammasome via AMPK/mTOR Autophagy Pathway
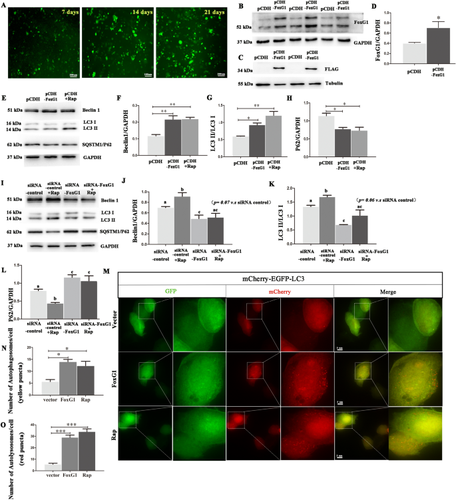
An increasing body of research suggests that promoting microglial autophagy hinders the neuroinflammation initiated though the NLRP3 inflammasome activation in Alzheimer’s disease (AD). The function of FoxG1, a crucial transcription factor involved in cell survival by regulating mitochondrial function, remains unknown during the AD process and neuroinflammation occurs. In the present study, we firstly found that Aβ peptides induced AD-like neuroinflammation upregulation and downregulated the level of autophagy. Following low-dose Aβ25–35 stimulation, FoxG1 expression and autophagy exhibited a gradual increase. Nevertheless, with high-concentration Aβ25–35 treatment, progressive decrease in FoxG1 expression and autophagy levels as the concentration of Aβ25–35 escalated. In addition, FoxG1 has a positive effect on cell viability and autophagy in the nervous system. In parallel with the Aβ25–35 stimulation, we employed siRNA to decrease the expression of FoxG1 in N2A cells. A substantial reduction in autophagy level (Beclin1, LC3II, SQSTM1/P62) and a notable growth in inflammatory response (NLRP3, TNF-α, and IL-6) were observed. In addition, we found FoxG1 overexpression owned the effect on the activation of AMPK/mTOR autophagy pathway and siRNA-FoxG1 successfully abolished this effect. Lastly, FoxG1 suppressed the NLRP3 inflammasome and enhanced the cognitive function in AD-like mouse model induced by Aβ25–35. Confirmed by cellular and animal experiments, FoxG1 suppressed NLRP3-mediated neuroinflammation, which was strongly linked to autophagy regulated by AMPK/mTOR. Taken together, FoxG1 may be a critical node in the pathologic progression of AD and has the potential to serve as therapeutic target.
期刊介绍:
Cellular and Molecular Neurobiology publishes original research concerned with the analysis of neuronal and brain function at the cellular and subcellular levels. The journal offers timely, peer-reviewed articles that describe anatomic, genetic, physiologic, pharmacologic, and biochemical approaches to the study of neuronal function and the analysis of elementary mechanisms. Studies are presented on isolated mammalian tissues and intact animals, with investigations aimed at the molecular mechanisms or neuronal responses at the level of single cells. Cellular and Molecular Neurobiology also presents studies of the effects of neurons on other organ systems, such as analysis of the electrical or biochemical response to neurotransmitters or neurohormones on smooth muscle or gland cells.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


