两个不同伊朗家族中与 RARS1 相关的骨髓营养不良性白质营养不良症-9 (HLD-9):病例报告和文献综述
IF 1.5
4区 医学
Q4 GENETICS & HEREDITY
引用次数: 0
摘要
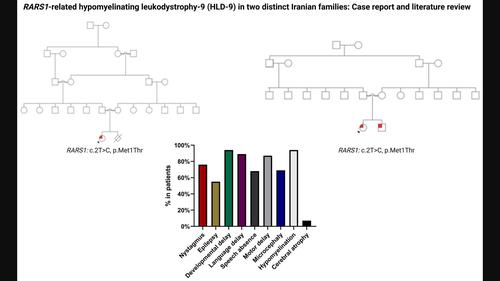
背景骨髓营养不良性白质营养不良-9(HLD-9)是由RARS1的双倍性致病变体引起的,RARS1编码细胞质精氨酸tRNA合成酶(ArgRS)。本研究旨在评估 RARS1 相关疾病患者的临床、神经放射学和遗传学特征,并确定基因型与表型之间的可能关系。结果在三名 HLD-9 患者中发现了 RARS1 同源变体(c.2T>C (p.Met1Thr))。所有患者的临床症状都很严重。经过文献回顾,我们从 8 项研究中找到了 30 例 HLD-9 病例。这 33 例患者的主要症状是骨髓营养不良、语言发育迟缓、智力障碍或发育迟缓。在已知发病年龄的 33 例 HLD9 患者中,平均发病年龄为 5.8 个月(SD = 8.1)。发病年龄的四分位数范围为 0-10 个月。结论 RARS1 中的致病变体会降低 ArgRS 的活性并导致多种症状,从伴有脑萎缩的严重早发性癫痫性脑病到伴有相对维持的髓鞘化的轻症。这些症状包括伴有眼球震颤和痉挛的典型髓鞘功能减退表现。此外,c.2T>C(p.Met1Thr)变异的致病性已经得到证实。本文章由计算机程序翻译,如有差异,请以英文原文为准。
RARS1‐related hypomyelinating leukodystrophy‐9 (HLD‐9) in two distinct Iranian families: Case report and literature review
BackgroundHypomyelinating leukodystrophy‐9 (HLD‐9) is caused by biallelic pathogenic variants in RARS1 , which codes for the cytoplasmic tRNA synthetase for arginine (ArgRS). This study aims to evaluate the clinical, neuroradiological, and genetic characteristics of patients with RARS1‐related disease and determine probable genotype–phenotype relationships.MethodsWe identified three patients with RARS1 homozygous pathogenic variants. Furthermore, we performed a comprehensive review of the literature.ResultsHomozygous variants of RARS1 (c.2T>C (p.Met1Thr)) were identified in three patients with HLD‐9. Clinical symptoms were severe in all patients. Following the literature review, thirty HLD‐9 cases from eight studies were found. The 33 patients' main symptoms were hypomyelination, language delay, and intellectual disability or developmental delay. The mean age of onset for HLD9 in the group of 33 patients with a known age of onset was 5.8 months (SD = 8.1). The interquartile range of age of onset was 0–10 months. Of the 25 variants identified, c.5A>G (p.Asp2Gly) was identified in 11 patients.ConclusionPathogenic variants in RARS1 decrease ArgRS activity and cause a wide range of symptoms, from severe, early onset epileptic encephalopathy with brain atrophy to a mild condition with relatively maintained myelination. These symptoms include the classic hypomyelination presentation with nystagmus and spasticity. Furthermore, the pathogenicity of the variation c.2T>C (p.Met1Thr) has been shown.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Molecular Genetics & Genomic Medicine
Biochemistry, Genetics and Molecular Biology-Genetics
CiteScore
4.20
自引率
0.00%
发文量
241
审稿时长
14 weeks
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


