Bernadette Mohr, Thor van Heesch, Alberto Pérez de Alba Ortíz, Jocelyne Vreede
{"title":"增强 DNA 分子模拟的采样策略","authors":"Bernadette Mohr, Thor van Heesch, Alberto Pérez de Alba Ortíz, Jocelyne Vreede","doi":"10.1002/wcms.1712","DOIUrl":null,"url":null,"abstract":"<p>Molecular dynamics (MD) simulations can provide detailed insights into complex molecular systems, such as DNA, at high resolution in space and time. Using current computer architectures, time scales of tens of microseconds are feasible with contemporary all-atom force fields. However, these timescales are insufficient to accurately characterize large conformational transitions in DNA and compare calculations to experimental data. This review discusses the advantages and drawbacks of two simulation approaches to overcome the timescale challenge. The first approach is based on adding biasing potentials to the system to drive transitions. Umbrella sampling, steered MD, and metadynamics are examples of these methods. A key challenge of such methods is the necessity of selecting one or a few efficient coordinates, commonly referred to as collective variables (CVs), along which to apply the biasing potential. The path-metadynamics methodology addresses this issue by finding the optimal route(s) between states in a multi-dimensional CV space. The second strategy is path sampling, which focuses MD simulations on the transitions. The assumption is that even though transitions between states are rare, they are generally fast. Stopping the simulations as soon as they reach a stable state can significantly increase simulation efficiency. We introduce these methods on the two-dimensional Müller–Brown potential. DNA applications are featured for two different processes: the Watson–Crick–Franklin to Hoogsteen transition in adenine–thymine base pairs and the binding of a DNA-binding protein domain to DNA.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 2","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2024-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1712","citationCount":"0","resultStr":"{\"title\":\"Enhanced sampling strategies for molecular simulation of DNA\",\"authors\":\"Bernadette Mohr, Thor van Heesch, Alberto Pérez de Alba Ortíz, Jocelyne Vreede\",\"doi\":\"10.1002/wcms.1712\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Molecular dynamics (MD) simulations can provide detailed insights into complex molecular systems, such as DNA, at high resolution in space and time. Using current computer architectures, time scales of tens of microseconds are feasible with contemporary all-atom force fields. However, these timescales are insufficient to accurately characterize large conformational transitions in DNA and compare calculations to experimental data. This review discusses the advantages and drawbacks of two simulation approaches to overcome the timescale challenge. The first approach is based on adding biasing potentials to the system to drive transitions. Umbrella sampling, steered MD, and metadynamics are examples of these methods. A key challenge of such methods is the necessity of selecting one or a few efficient coordinates, commonly referred to as collective variables (CVs), along which to apply the biasing potential. The path-metadynamics methodology addresses this issue by finding the optimal route(s) between states in a multi-dimensional CV space. The second strategy is path sampling, which focuses MD simulations on the transitions. The assumption is that even though transitions between states are rare, they are generally fast. Stopping the simulations as soon as they reach a stable state can significantly increase simulation efficiency. We introduce these methods on the two-dimensional Müller–Brown potential. DNA applications are featured for two different processes: the Watson–Crick–Franklin to Hoogsteen transition in adenine–thymine base pairs and the binding of a DNA-binding protein domain to DNA.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 2\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2024-04-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1712\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1712\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1712","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
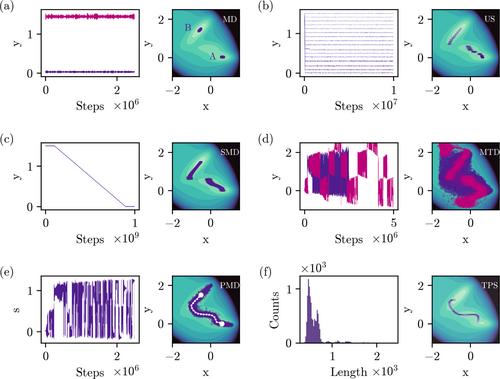
分子动力学(MD)模拟可以在空间和时间上以高分辨率详细了解 DNA 等复杂分子系统。利用当前的计算机架构,几十微秒的时间尺度在当代全原子力场中是可行的。然而,这些时间尺度不足以准确描述 DNA 中的大型构象转变,也不足以将计算结果与实验数据进行比较。本综述讨论了克服时间尺度挑战的两种模拟方法的优缺点。第一种方法基于在系统中添加偏置电位来驱动转变。伞状采样、定向 MD 和元动力学就是这些方法的例子。这些方法面临的一个主要挑战是,必须选择一个或几个有效坐标(通常称为集体变量(CV))来应用偏置电势。路径计量学方法通过在多维 CV 空间中寻找状态之间的最佳路径来解决这一问题。第二种策略是路径采样,它将 MD 模拟的重点放在转换上。我们的假设是,尽管状态之间的转换很少,但转换速度通常很快。一旦达到稳定状态,立即停止模拟,可以显著提高模拟效率。我们在二维 Müller-Brown 势上介绍了这些方法。DNA 应用是两个不同过程的特色:腺嘌呤-胸腺嘧啶碱基对中沃森-克里克-弗兰克林到霍格斯坦的转变,以及 DNA 结合蛋白结构域与 DNA 的结合:
Enhanced sampling strategies for molecular simulation of DNA
Molecular dynamics (MD) simulations can provide detailed insights into complex molecular systems, such as DNA, at high resolution in space and time. Using current computer architectures, time scales of tens of microseconds are feasible with contemporary all-atom force fields. However, these timescales are insufficient to accurately characterize large conformational transitions in DNA and compare calculations to experimental data. This review discusses the advantages and drawbacks of two simulation approaches to overcome the timescale challenge. The first approach is based on adding biasing potentials to the system to drive transitions. Umbrella sampling, steered MD, and metadynamics are examples of these methods. A key challenge of such methods is the necessity of selecting one or a few efficient coordinates, commonly referred to as collective variables (CVs), along which to apply the biasing potential. The path-metadynamics methodology addresses this issue by finding the optimal route(s) between states in a multi-dimensional CV space. The second strategy is path sampling, which focuses MD simulations on the transitions. The assumption is that even though transitions between states are rare, they are generally fast. Stopping the simulations as soon as they reach a stable state can significantly increase simulation efficiency. We introduce these methods on the two-dimensional Müller–Brown potential. DNA applications are featured for two different processes: the Watson–Crick–Franklin to Hoogsteen transition in adenine–thymine base pairs and the binding of a DNA-binding protein domain to DNA.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


