Ying Chen, Run Yang, Xin Chen, Naier Lin, Chenlong Li, Yaoyao Fu, Aijuan He, Yimin Wang, Tianyu Zhang, Jing Ma
{"title":"通过鉴定 EFTUD2 的新型致病突变,诊断出伴有小头畸形的非典型下颌骨面部发育不良症。","authors":"Ying Chen, Run Yang, Xin Chen, Naier Lin, Chenlong Li, Yaoyao Fu, Aijuan He, Yimin Wang, Tianyu Zhang, Jing Ma","doi":"10.1002/mgg3.2426","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mandibulofacial dysostosis with microcephaly (MFDM, OMIM# 610536) is a rare monogenic disease that is caused by a mutation in the elongation factor Tu GTP binding domain containing 2 gene (EFTUD2, OMIM* 603892). It is characterized by mandibulofacial dysplasia, microcephaly, malformed ears, cleft palate, growth and intellectual disability. MFDM can be easily misdiagnosed due to its phenotypic overlap with other craniofacial dysostosis syndromes. The clinical presentation of MFDM is highly variable among patients.</p><p><strong>Methods: </strong>A patient with craniofacial anomalies was enrolled and evaluated by a multidisciplinary team. To make a definitive diagnosis, whole-exome sequencing was performed, followed by validation by Sanger sequencing.</p><p><strong>Results: </strong>The patient presented with extensive facial bone dysostosis, upward slanting palpebral fissures, outer and middle ear malformation, a previously unreported orbit anomaly, and spina bifida occulta. A novel, pathogenic insertion mutation (c.215_216insT: p.Tyr73Valfs*4) in EFTUD2 was identified as the likely cause of the disease.</p><p><strong>Conclusions: </strong>We diagnosed this atypical case of MFDM by the detection of a novel pathogenetic mutation in EFTUD2. We also observed previously unreported features. These findings enrich both the genotypic and phenotypic spectrum of MFDM.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":null,"pages":null},"PeriodicalIF":1.5000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10985408/pdf/","citationCount":"0","resultStr":"{\"title\":\"Atypical mandibulofacial dysostosis with microcephaly diagnosed through the identification of a novel pathogenic mutation in EFTUD2.\",\"authors\":\"Ying Chen, Run Yang, Xin Chen, Naier Lin, Chenlong Li, Yaoyao Fu, Aijuan He, Yimin Wang, Tianyu Zhang, Jing Ma\",\"doi\":\"10.1002/mgg3.2426\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mandibulofacial dysostosis with microcephaly (MFDM, OMIM# 610536) is a rare monogenic disease that is caused by a mutation in the elongation factor Tu GTP binding domain containing 2 gene (EFTUD2, OMIM* 603892). It is characterized by mandibulofacial dysplasia, microcephaly, malformed ears, cleft palate, growth and intellectual disability. MFDM can be easily misdiagnosed due to its phenotypic overlap with other craniofacial dysostosis syndromes. The clinical presentation of MFDM is highly variable among patients.</p><p><strong>Methods: </strong>A patient with craniofacial anomalies was enrolled and evaluated by a multidisciplinary team. To make a definitive diagnosis, whole-exome sequencing was performed, followed by validation by Sanger sequencing.</p><p><strong>Results: </strong>The patient presented with extensive facial bone dysostosis, upward slanting palpebral fissures, outer and middle ear malformation, a previously unreported orbit anomaly, and spina bifida occulta. A novel, pathogenic insertion mutation (c.215_216insT: p.Tyr73Valfs*4) in EFTUD2 was identified as the likely cause of the disease.</p><p><strong>Conclusions: </strong>We diagnosed this atypical case of MFDM by the detection of a novel pathogenetic mutation in EFTUD2. We also observed previously unreported features. These findings enrich both the genotypic and phenotypic spectrum of MFDM.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10985408/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.2426\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2426","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Atypical mandibulofacial dysostosis with microcephaly diagnosed through the identification of a novel pathogenic mutation in EFTUD2.
Background: Mandibulofacial dysostosis with microcephaly (MFDM, OMIM# 610536) is a rare monogenic disease that is caused by a mutation in the elongation factor Tu GTP binding domain containing 2 gene (EFTUD2, OMIM* 603892). It is characterized by mandibulofacial dysplasia, microcephaly, malformed ears, cleft palate, growth and intellectual disability. MFDM can be easily misdiagnosed due to its phenotypic overlap with other craniofacial dysostosis syndromes. The clinical presentation of MFDM is highly variable among patients.
Methods: A patient with craniofacial anomalies was enrolled and evaluated by a multidisciplinary team. To make a definitive diagnosis, whole-exome sequencing was performed, followed by validation by Sanger sequencing.
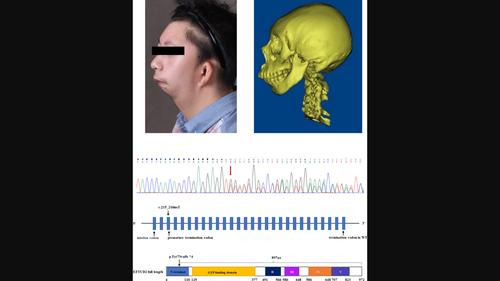
Results: The patient presented with extensive facial bone dysostosis, upward slanting palpebral fissures, outer and middle ear malformation, a previously unreported orbit anomaly, and spina bifida occulta. A novel, pathogenic insertion mutation (c.215_216insT: p.Tyr73Valfs*4) in EFTUD2 was identified as the likely cause of the disease.
Conclusions: We diagnosed this atypical case of MFDM by the detection of a novel pathogenetic mutation in EFTUD2. We also observed previously unreported features. These findings enrich both the genotypic and phenotypic spectrum of MFDM.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


