{"title":"大剂量氨溴索治疗神经病变性戈谢病疗效迅速而持久:病例报告和文献综述。","authors":"Kanako Higashi, Yuri Sonoda, Noriyuki Kaku, Fumihiko Fujii, Fumiya Yamashita, Sooyoung Lee, Vlad Tocan, Go Ebihara, Wakato Matsuoka, Kenichi Tetsuhara, Motoshi Sonoda, Pin Fee Chong, Yuichi Mushimoto, Kanako Kojima-Ishii, Masataka Ishimura, Yuhki Koga, Atsuhisa Fukuta, Nana Akagi Tsuchihashi, Yoshikazu Kikuchi, Takahito Karashima, Takaaki Sawada, Taeko Hotta, Makoto Yoshimitsu, Hideyuki Terazono, Tatsuro Tajiri, Takashi Nakagawa, Yasunari Sakai, Kimitoshi Nakamura, Shouichi Ohga","doi":"10.1002/mgg3.2427","DOIUrl":null,"url":null,"abstract":"<p><p>Gaucher disease (GD) is a lysosomal storage disorder caused by a deficiency in the GBA1-encoded enzyme, β-glucocerebrosidase. Enzyme replacement therapy is ineffective for neuronopathic Gaucher disease (nGD). High-dose ambroxol has been administered as an alternative treatment for a group of patients with nGD. However, little is known about the clinical indication and the long-term outcome of patients after ambroxol therapy. We herein report a case of a female patient who presented with a progressive disease of GD type 2 from 11 months of age and had the pathogenic variants of p.L483P (formerly defined as p.L444P) and p.R502H (p.R463H) in GBA1. A combined treatment of imiglucerase with ambroxol started improving the patient's motor activity in 1 week, while it kept the long-lasting effect of preventing the deteriorating phenotype for 30 months. A literature review identified 40 patients with nGD, who had received high-dose ambroxol therapy. More than 65% of these patients favorably responded to the molecular chaperone therapy, irrespective of p.L483P homozygous, heterozygous or the other genotypes. These results highlight the long-lasting effect of ambroxol-based chaperone therapy for patients with an expanding spectrum of mutations in GBA1.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":null,"pages":null},"PeriodicalIF":1.5000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10980885/pdf/","citationCount":"0","resultStr":"{\"title\":\"Rapid and long-lasting efficacy of high-dose ambroxol therapy for neuronopathic Gaucher disease: A case report and literature review.\",\"authors\":\"Kanako Higashi, Yuri Sonoda, Noriyuki Kaku, Fumihiko Fujii, Fumiya Yamashita, Sooyoung Lee, Vlad Tocan, Go Ebihara, Wakato Matsuoka, Kenichi Tetsuhara, Motoshi Sonoda, Pin Fee Chong, Yuichi Mushimoto, Kanako Kojima-Ishii, Masataka Ishimura, Yuhki Koga, Atsuhisa Fukuta, Nana Akagi Tsuchihashi, Yoshikazu Kikuchi, Takahito Karashima, Takaaki Sawada, Taeko Hotta, Makoto Yoshimitsu, Hideyuki Terazono, Tatsuro Tajiri, Takashi Nakagawa, Yasunari Sakai, Kimitoshi Nakamura, Shouichi Ohga\",\"doi\":\"10.1002/mgg3.2427\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Gaucher disease (GD) is a lysosomal storage disorder caused by a deficiency in the GBA1-encoded enzyme, β-glucocerebrosidase. Enzyme replacement therapy is ineffective for neuronopathic Gaucher disease (nGD). High-dose ambroxol has been administered as an alternative treatment for a group of patients with nGD. However, little is known about the clinical indication and the long-term outcome of patients after ambroxol therapy. We herein report a case of a female patient who presented with a progressive disease of GD type 2 from 11 months of age and had the pathogenic variants of p.L483P (formerly defined as p.L444P) and p.R502H (p.R463H) in GBA1. A combined treatment of imiglucerase with ambroxol started improving the patient's motor activity in 1 week, while it kept the long-lasting effect of preventing the deteriorating phenotype for 30 months. A literature review identified 40 patients with nGD, who had received high-dose ambroxol therapy. More than 65% of these patients favorably responded to the molecular chaperone therapy, irrespective of p.L483P homozygous, heterozygous or the other genotypes. These results highlight the long-lasting effect of ambroxol-based chaperone therapy for patients with an expanding spectrum of mutations in GBA1.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10980885/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.2427\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2427","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Rapid and long-lasting efficacy of high-dose ambroxol therapy for neuronopathic Gaucher disease: A case report and literature review.
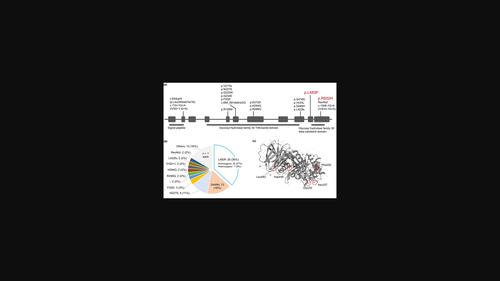
Gaucher disease (GD) is a lysosomal storage disorder caused by a deficiency in the GBA1-encoded enzyme, β-glucocerebrosidase. Enzyme replacement therapy is ineffective for neuronopathic Gaucher disease (nGD). High-dose ambroxol has been administered as an alternative treatment for a group of patients with nGD. However, little is known about the clinical indication and the long-term outcome of patients after ambroxol therapy. We herein report a case of a female patient who presented with a progressive disease of GD type 2 from 11 months of age and had the pathogenic variants of p.L483P (formerly defined as p.L444P) and p.R502H (p.R463H) in GBA1. A combined treatment of imiglucerase with ambroxol started improving the patient's motor activity in 1 week, while it kept the long-lasting effect of preventing the deteriorating phenotype for 30 months. A literature review identified 40 patients with nGD, who had received high-dose ambroxol therapy. More than 65% of these patients favorably responded to the molecular chaperone therapy, irrespective of p.L483P homozygous, heterozygous or the other genotypes. These results highlight the long-lasting effect of ambroxol-based chaperone therapy for patients with an expanding spectrum of mutations in GBA1.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


