{"title":"通过全外显子组测序和 LC-MS/MS 重新分析确诊的磷酸丝氨酸氨基转移酶缺乏症:病例报告和文献综述。","authors":"Jiaci Li, Xinping Wei, Yuchen Sun, Xiaofang Chen, Ying Zhang, Xiaoyu Cui, Jianbo Shu, Dong Li, Chunquan Cai","doi":"10.1002/mgg3.2400","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Phosphoserine aminotransferase deficiency (PSATD) is an autosomal recessive disorder associated with hypertonia, psychomotor retardation, and acquired microcephaly. Patients with PSATD have low concentrations of serine in plasma and cerebrospinal fluid.</p><p><strong>Methods: </strong>We reported a 2-year-old female child with developmental delay, dyskinesia, and microcephaly. LC-MS/MS was used to detect amino acid concentration in the blood and whole-exome sequencing (WES) was used to identify the variants. PolyPhen-2 web server and PyMol were used to predict the pathogenicity and changes in the 3D model molecular structure of protein caused by variants.</p><p><strong>Results: </strong>WES demonstrated compound heterozygous variants in PSAT1, which is associated with PSATD, with a paternal likely pathogenic variant (c.235G>A, Gly79Arg) and a maternal likely pathogenic variant (c.43G>C, Ala15Pro). Reduced serine concentration in LC-MS/MS further confirmed the diagnosis of PSATD in this patient.</p><p><strong>Conclusions: </strong>Our findings demonstrate the importance of WES combined with LC-MS/MS reanalysis in the diagnosis of genetic diseases and expand the PSAT1 variant spectrum in PSATD. Moreover, we summarize all the cases caused by PSAT1 variants in the literature. This case provides a vital reference for the diagnosis of future cases.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":null,"pages":null},"PeriodicalIF":1.5000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10976427/pdf/","citationCount":"0","resultStr":"{\"title\":\"Phosphoserine aminotransferase deficiency diagnosed by whole-exome sequencing and LC-MS/MS reanalysis: A case report and review of literature.\",\"authors\":\"Jiaci Li, Xinping Wei, Yuchen Sun, Xiaofang Chen, Ying Zhang, Xiaoyu Cui, Jianbo Shu, Dong Li, Chunquan Cai\",\"doi\":\"10.1002/mgg3.2400\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Phosphoserine aminotransferase deficiency (PSATD) is an autosomal recessive disorder associated with hypertonia, psychomotor retardation, and acquired microcephaly. Patients with PSATD have low concentrations of serine in plasma and cerebrospinal fluid.</p><p><strong>Methods: </strong>We reported a 2-year-old female child with developmental delay, dyskinesia, and microcephaly. LC-MS/MS was used to detect amino acid concentration in the blood and whole-exome sequencing (WES) was used to identify the variants. PolyPhen-2 web server and PyMol were used to predict the pathogenicity and changes in the 3D model molecular structure of protein caused by variants.</p><p><strong>Results: </strong>WES demonstrated compound heterozygous variants in PSAT1, which is associated with PSATD, with a paternal likely pathogenic variant (c.235G>A, Gly79Arg) and a maternal likely pathogenic variant (c.43G>C, Ala15Pro). Reduced serine concentration in LC-MS/MS further confirmed the diagnosis of PSATD in this patient.</p><p><strong>Conclusions: </strong>Our findings demonstrate the importance of WES combined with LC-MS/MS reanalysis in the diagnosis of genetic diseases and expand the PSAT1 variant spectrum in PSATD. Moreover, we summarize all the cases caused by PSAT1 variants in the literature. This case provides a vital reference for the diagnosis of future cases.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10976427/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.2400\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2400","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Phosphoserine aminotransferase deficiency diagnosed by whole-exome sequencing and LC-MS/MS reanalysis: A case report and review of literature.
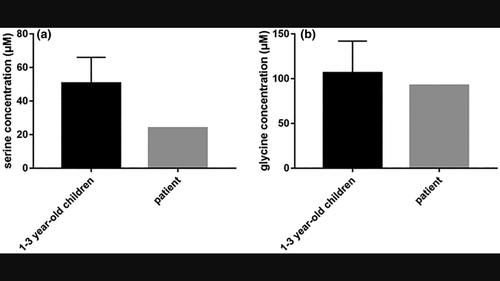
Background: Phosphoserine aminotransferase deficiency (PSATD) is an autosomal recessive disorder associated with hypertonia, psychomotor retardation, and acquired microcephaly. Patients with PSATD have low concentrations of serine in plasma and cerebrospinal fluid.
Methods: We reported a 2-year-old female child with developmental delay, dyskinesia, and microcephaly. LC-MS/MS was used to detect amino acid concentration in the blood and whole-exome sequencing (WES) was used to identify the variants. PolyPhen-2 web server and PyMol were used to predict the pathogenicity and changes in the 3D model molecular structure of protein caused by variants.
Results: WES demonstrated compound heterozygous variants in PSAT1, which is associated with PSATD, with a paternal likely pathogenic variant (c.235G>A, Gly79Arg) and a maternal likely pathogenic variant (c.43G>C, Ala15Pro). Reduced serine concentration in LC-MS/MS further confirmed the diagnosis of PSATD in this patient.
Conclusions: Our findings demonstrate the importance of WES combined with LC-MS/MS reanalysis in the diagnosis of genetic diseases and expand the PSAT1 variant spectrum in PSATD. Moreover, we summarize all the cases caused by PSAT1 variants in the literature. This case provides a vital reference for the diagnosis of future cases.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


