Rebecca Bruns, Khurram Liaqat, Abdul Nasir, Kayla Treat, Vinaya S. Murthy, Lili Mantcheva, Wilfredo Torres, Erin Conboy, Francesco Vetrini
{"title":"未确诊罕见病诊所在两姐妹安杰尔曼综合征患者中发现新型 UBE3A 变异:诊断奥德赛的终点。","authors":"Rebecca Bruns, Khurram Liaqat, Abdul Nasir, Kayla Treat, Vinaya S. Murthy, Lili Mantcheva, Wilfredo Torres, Erin Conboy, Francesco Vetrini","doi":"10.1111/cga.12566","DOIUrl":null,"url":null,"abstract":"<p>Angelman syndrome (AS, MIM #105830) is a neurodevelopmental disorder characterized by severe intellectual disability, profound developmental delay, movement or balance problems, an excessively cheerful disposition, and seizures. AS results from inadequate expression of the maternal <i>UBE3A</i> gene (MIM #601623), which encodes an E3 ligase in the ubiquitin-proteasome pathway. Here we present the case of two sisters with features consistent with AS who had negative methylation analyses. An autism/intellectual disability expanded panel revealed a maternally inherited novel <i>UBE3A</i> (NM_001354506.2) variant c.2443C>T p.(Pro815Ser) in both patients that was initially classified as a variant of uncertain significance. The patients were enrolled in Indiana University's Undiagnosed Rare Disease Clinic (URDC) to further investigate the variant. Additional data, including deep phenotyping, familial segregation analysis, and in silico studies, suggest that the variant is likely pathogenic. 3D modeling studies based on the available crystal structure revealed that the Pro815Ser variant can introduce more flexibility into the protein and alter its enzymatic activity. Recent literature confirms the pathogenic nature of the variant. Reanalysis of the <i>UBE3A</i> variant has heightened existing knowledge of AS and has offered this family an end to their diagnostic odyssey.</p>","PeriodicalId":10626,"journal":{"name":"Congenital Anomalies","volume":"64 3","pages":"155-160"},"PeriodicalIF":1.6000,"publicationDate":"2024-03-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cga.12566","citationCount":"0","resultStr":"{\"title\":\"Undiagnosed rare disease clinic identifies a novel UBE3A variant in two sisters with Angelman syndrome: The end of a diagnostic odyssey\",\"authors\":\"Rebecca Bruns, Khurram Liaqat, Abdul Nasir, Kayla Treat, Vinaya S. Murthy, Lili Mantcheva, Wilfredo Torres, Erin Conboy, Francesco Vetrini\",\"doi\":\"10.1111/cga.12566\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Angelman syndrome (AS, MIM #105830) is a neurodevelopmental disorder characterized by severe intellectual disability, profound developmental delay, movement or balance problems, an excessively cheerful disposition, and seizures. AS results from inadequate expression of the maternal <i>UBE3A</i> gene (MIM #601623), which encodes an E3 ligase in the ubiquitin-proteasome pathway. Here we present the case of two sisters with features consistent with AS who had negative methylation analyses. An autism/intellectual disability expanded panel revealed a maternally inherited novel <i>UBE3A</i> (NM_001354506.2) variant c.2443C>T p.(Pro815Ser) in both patients that was initially classified as a variant of uncertain significance. The patients were enrolled in Indiana University's Undiagnosed Rare Disease Clinic (URDC) to further investigate the variant. Additional data, including deep phenotyping, familial segregation analysis, and in silico studies, suggest that the variant is likely pathogenic. 3D modeling studies based on the available crystal structure revealed that the Pro815Ser variant can introduce more flexibility into the protein and alter its enzymatic activity. Recent literature confirms the pathogenic nature of the variant. Reanalysis of the <i>UBE3A</i> variant has heightened existing knowledge of AS and has offered this family an end to their diagnostic odyssey.</p>\",\"PeriodicalId\":10626,\"journal\":{\"name\":\"Congenital Anomalies\",\"volume\":\"64 3\",\"pages\":\"155-160\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2024-03-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/cga.12566\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Congenital Anomalies\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/cga.12566\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Congenital Anomalies","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cga.12566","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PEDIATRICS","Score":null,"Total":0}
Undiagnosed rare disease clinic identifies a novel UBE3A variant in two sisters with Angelman syndrome: The end of a diagnostic odyssey
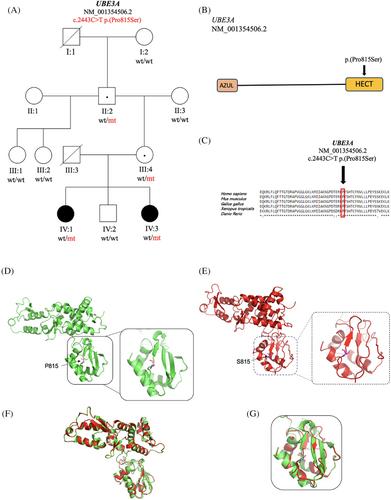
Angelman syndrome (AS, MIM #105830) is a neurodevelopmental disorder characterized by severe intellectual disability, profound developmental delay, movement or balance problems, an excessively cheerful disposition, and seizures. AS results from inadequate expression of the maternal UBE3A gene (MIM #601623), which encodes an E3 ligase in the ubiquitin-proteasome pathway. Here we present the case of two sisters with features consistent with AS who had negative methylation analyses. An autism/intellectual disability expanded panel revealed a maternally inherited novel UBE3A (NM_001354506.2) variant c.2443C>T p.(Pro815Ser) in both patients that was initially classified as a variant of uncertain significance. The patients were enrolled in Indiana University's Undiagnosed Rare Disease Clinic (URDC) to further investigate the variant. Additional data, including deep phenotyping, familial segregation analysis, and in silico studies, suggest that the variant is likely pathogenic. 3D modeling studies based on the available crystal structure revealed that the Pro815Ser variant can introduce more flexibility into the protein and alter its enzymatic activity. Recent literature confirms the pathogenic nature of the variant. Reanalysis of the UBE3A variant has heightened existing knowledge of AS and has offered this family an end to their diagnostic odyssey.
期刊介绍:
Congenital Anomalies is the official English language journal of the Japanese Teratology Society, and publishes original articles in laboratory as well as clinical research in all areas of abnormal development and related fields, from all over the world. Although contributions by members of the teratology societies affiliated with The International Federation of Teratology Societies are given priority, contributions from non-members are welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


