Huijuan Chen , Bing Wang , Lili Cai , Xiaotian Yang , Yali Hu , Yiran Zhang , Xue Leng , Wen Liu , Dongjie Fan , Beifang Niu , Qiming Zhou
{"title":"癌症新一代测序分析中检测和估计人类样本交叉污染的计算方法的综合性能评估、比较和整合。","authors":"Huijuan Chen , Bing Wang , Lili Cai , Xiaotian Yang , Yali Hu , Yiran Zhang , Xue Leng , Wen Liu , Dongjie Fan , Beifang Niu , Qiming Zhou","doi":"10.1016/j.jbi.2024.104625","DOIUrl":null,"url":null,"abstract":"<div><p>Cross-sample contamination is one of the major issues in next-generation sequencing (NGS)-based molecular assays. This type of contamination, even at very low levels, can significantly impact the results of an analysis, especially in the detection of somatic alterations in tumor samples. Several contamination identification tools have been developed and implemented as a crucial quality-control step in the routine NGS bioinformatic pipeline. However, no study has been published to comprehensively and systematically investigate, evaluate, and compare these computational methods in the cancer NGS analysis. In this study, we comprehensively investigated nine state-of-the-art computational methods for detecting cross-sample contamination. To explore their application in cancer NGS analysis, we further compared the performance of five representative tools by qualitative and quantitative analyses using <em>in silico</em> and simulated experimental NGS data. The results showed that Conpair achieved the best performance for identifying contamination and predicting the level of contamination in solid tumors NGS analysis. Moreover, based on Conpair, we developed a Python script, Contamination Source Predictor (ConSPr), to identify the source of contamination. We anticipate that this comprehensive survey and the proposed tool for predicting the source of contamination will assist researchers in selecting appropriate cross-contamination detection tools in cancer NGS analysis and inspire the development of computational methods for detecting sample cross-contamination and identifying its source in the future.</p></div>","PeriodicalId":15263,"journal":{"name":"Journal of Biomedical Informatics","volume":null,"pages":null},"PeriodicalIF":4.0000,"publicationDate":"2024-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A comprehensive performance evaluation, comparison, and integration of computational methods for detecting and estimating cross-contamination of human samples in cancer next-generation sequencing analysis\",\"authors\":\"Huijuan Chen , Bing Wang , Lili Cai , Xiaotian Yang , Yali Hu , Yiran Zhang , Xue Leng , Wen Liu , Dongjie Fan , Beifang Niu , Qiming Zhou\",\"doi\":\"10.1016/j.jbi.2024.104625\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Cross-sample contamination is one of the major issues in next-generation sequencing (NGS)-based molecular assays. This type of contamination, even at very low levels, can significantly impact the results of an analysis, especially in the detection of somatic alterations in tumor samples. Several contamination identification tools have been developed and implemented as a crucial quality-control step in the routine NGS bioinformatic pipeline. However, no study has been published to comprehensively and systematically investigate, evaluate, and compare these computational methods in the cancer NGS analysis. In this study, we comprehensively investigated nine state-of-the-art computational methods for detecting cross-sample contamination. To explore their application in cancer NGS analysis, we further compared the performance of five representative tools by qualitative and quantitative analyses using <em>in silico</em> and simulated experimental NGS data. The results showed that Conpair achieved the best performance for identifying contamination and predicting the level of contamination in solid tumors NGS analysis. Moreover, based on Conpair, we developed a Python script, Contamination Source Predictor (ConSPr), to identify the source of contamination. We anticipate that this comprehensive survey and the proposed tool for predicting the source of contamination will assist researchers in selecting appropriate cross-contamination detection tools in cancer NGS analysis and inspire the development of computational methods for detecting sample cross-contamination and identifying its source in the future.</p></div>\",\"PeriodicalId\":15263,\"journal\":{\"name\":\"Journal of Biomedical Informatics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2024-03-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Biomedical Informatics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1532046424000431\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Biomedical Informatics","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1532046424000431","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
A comprehensive performance evaluation, comparison, and integration of computational methods for detecting and estimating cross-contamination of human samples in cancer next-generation sequencing analysis
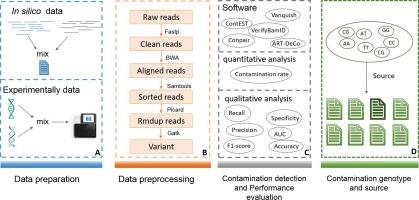
Cross-sample contamination is one of the major issues in next-generation sequencing (NGS)-based molecular assays. This type of contamination, even at very low levels, can significantly impact the results of an analysis, especially in the detection of somatic alterations in tumor samples. Several contamination identification tools have been developed and implemented as a crucial quality-control step in the routine NGS bioinformatic pipeline. However, no study has been published to comprehensively and systematically investigate, evaluate, and compare these computational methods in the cancer NGS analysis. In this study, we comprehensively investigated nine state-of-the-art computational methods for detecting cross-sample contamination. To explore their application in cancer NGS analysis, we further compared the performance of five representative tools by qualitative and quantitative analyses using in silico and simulated experimental NGS data. The results showed that Conpair achieved the best performance for identifying contamination and predicting the level of contamination in solid tumors NGS analysis. Moreover, based on Conpair, we developed a Python script, Contamination Source Predictor (ConSPr), to identify the source of contamination. We anticipate that this comprehensive survey and the proposed tool for predicting the source of contamination will assist researchers in selecting appropriate cross-contamination detection tools in cancer NGS analysis and inspire the development of computational methods for detecting sample cross-contamination and identifying its source in the future.
期刊介绍:
The Journal of Biomedical Informatics reflects a commitment to high-quality original research papers, reviews, and commentaries in the area of biomedical informatics methodology. Although we publish articles motivated by applications in the biomedical sciences (for example, clinical medicine, health care, population health, and translational bioinformatics), the journal emphasizes reports of new methodologies and techniques that have general applicability and that form the basis for the evolving science of biomedical informatics. Articles on medical devices; evaluations of implemented systems (including clinical trials of information technologies); or papers that provide insight into a biological process, a specific disease, or treatment options would generally be more suitable for publication in other venues. Papers on applications of signal processing and image analysis are often more suitable for biomedical engineering journals or other informatics journals, although we do publish papers that emphasize the information management and knowledge representation/modeling issues that arise in the storage and use of biological signals and images. System descriptions are welcome if they illustrate and substantiate the underlying methodology that is the principal focus of the report and an effort is made to address the generalizability and/or range of application of that methodology. Note also that, given the international nature of JBI, papers that deal with specific languages other than English, or with country-specific health systems or approaches, are acceptable for JBI only if they offer generalizable lessons that are relevant to the broad JBI readership, regardless of their country, language, culture, or health system.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


