Huy Hoang Nguyen Vo , Thu Huong Thi Phung , Khanh Linh Chung , Thien Y. Vu
{"title":"利用配体设计器精确切割,定制铬烯苯基 COX 抑制剂","authors":"Huy Hoang Nguyen Vo , Thu Huong Thi Phung , Khanh Linh Chung , Thien Y. Vu","doi":"10.1016/j.jmgm.2024.108747","DOIUrl":null,"url":null,"abstract":"<div><p>Cyclooxygenases 1 and 2 (COX-1/2) are enzymes renowned for inducing inflammatory responses through the production of prostaglandins. Thus, the development of COX inhibitors has been a promising approach for identifying compounds with anti-inflammatory potential. In this study, we designed 27 new compounds (<strong>1–27</strong>) based on the structure of a previously known COX inhibitor, using the Ligand Designer tool. Our aim was to improve the affinity of the compounds with COX enzymes by inducing interactions with residue Arg120 while retaining the good π-π stacking interactions of the chromene-phenyl scaffold. Through screening based on ligand-binding free energy defined by molecular docking simulations and MM/GBSA technique, compounds <strong>9</strong> and <strong>10</strong> were identified as having the highest ability to inhibit COX proteins. The binding affinities of the two compounds with COX-1/2 were superior to those of the original <strong>NAI10</strong> compound and the reference drug indomethacin. Our virtual screening suggests that compounds <strong>9</strong> and <strong>10</strong> have a strong ability to inhibit COX-1/2 and thus could be promising candidates for further anti-inflammatory drug studies. In essence, our study underscores the pivotal role of the N-aryl iminocoumarin scaffold in shaping the future landscape of novel anti-inflammatory drug development.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Precise cuts for tailoring chromene-phenyl COX inhibitors with Ligand Designer\",\"authors\":\"Huy Hoang Nguyen Vo , Thu Huong Thi Phung , Khanh Linh Chung , Thien Y. Vu\",\"doi\":\"10.1016/j.jmgm.2024.108747\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Cyclooxygenases 1 and 2 (COX-1/2) are enzymes renowned for inducing inflammatory responses through the production of prostaglandins. Thus, the development of COX inhibitors has been a promising approach for identifying compounds with anti-inflammatory potential. In this study, we designed 27 new compounds (<strong>1–27</strong>) based on the structure of a previously known COX inhibitor, using the Ligand Designer tool. Our aim was to improve the affinity of the compounds with COX enzymes by inducing interactions with residue Arg120 while retaining the good π-π stacking interactions of the chromene-phenyl scaffold. Through screening based on ligand-binding free energy defined by molecular docking simulations and MM/GBSA technique, compounds <strong>9</strong> and <strong>10</strong> were identified as having the highest ability to inhibit COX proteins. The binding affinities of the two compounds with COX-1/2 were superior to those of the original <strong>NAI10</strong> compound and the reference drug indomethacin. Our virtual screening suggests that compounds <strong>9</strong> and <strong>10</strong> have a strong ability to inhibit COX-1/2 and thus could be promising candidates for further anti-inflammatory drug studies. In essence, our study underscores the pivotal role of the N-aryl iminocoumarin scaffold in shaping the future landscape of novel anti-inflammatory drug development.</p></div>\",\"PeriodicalId\":16361,\"journal\":{\"name\":\"Journal of molecular graphics & modelling\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular graphics & modelling\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1093326324000470\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324000470","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Precise cuts for tailoring chromene-phenyl COX inhibitors with Ligand Designer
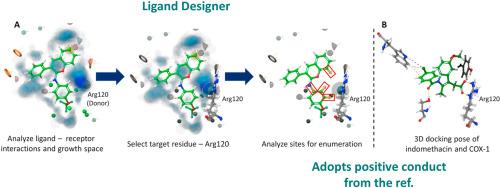
Cyclooxygenases 1 and 2 (COX-1/2) are enzymes renowned for inducing inflammatory responses through the production of prostaglandins. Thus, the development of COX inhibitors has been a promising approach for identifying compounds with anti-inflammatory potential. In this study, we designed 27 new compounds (1–27) based on the structure of a previously known COX inhibitor, using the Ligand Designer tool. Our aim was to improve the affinity of the compounds with COX enzymes by inducing interactions with residue Arg120 while retaining the good π-π stacking interactions of the chromene-phenyl scaffold. Through screening based on ligand-binding free energy defined by molecular docking simulations and MM/GBSA technique, compounds 9 and 10 were identified as having the highest ability to inhibit COX proteins. The binding affinities of the two compounds with COX-1/2 were superior to those of the original NAI10 compound and the reference drug indomethacin. Our virtual screening suggests that compounds 9 and 10 have a strong ability to inhibit COX-1/2 and thus could be promising candidates for further anti-inflammatory drug studies. In essence, our study underscores the pivotal role of the N-aryl iminocoumarin scaffold in shaping the future landscape of novel anti-inflammatory drug development.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


