{"title":"单核苷酸多态性标记对杂合度的估计取决于具体情况,而且往往是错误的。","authors":"Jarrod Sopniewski, Renee A. Catullo","doi":"10.1111/1755-0998.13947","DOIUrl":null,"url":null,"abstract":"<p>Genetic diversity is frequently described using heterozygosity, particularly in a conservation context. Often, it is estimated using single nucleotide polymorphisms (SNPs); however, it has been shown that heterozygosity values calculated from SNPs can be biased by both study design and filtering parameters. Though solutions have been proposed to address these issues, our own work has found them to be inadequate in some circumstances. Here, we aimed to improve the reliability and comparability of heterozygosity estimates, specifically by investigating how sample size and missing data thresholds influenced the calculation of autosomal heterozygosity (heterozygosity calculated from across the genome, i.e. fixed and variable sites). We also explored how the standard practice of tri- and tetra-allelic site exclusion could bias heterozygosity estimates and influence eventual conclusions relating to genetic diversity. Across three distinct taxa (a frog, <i>Litoria rubella</i>; a tree, <i>Eucalyptus microcarpa</i>; and a grasshopper, <i>Keyacris scurra</i>), we found heterozygosity estimates to be meaningfully affected by sample size and missing data thresholds, partly due to the exclusion of tri- and tetra-allelic sites. These biases were inconsistent both between species and populations, with more diverse populations tending to have their estimates more severely affected, thus having potential to dramatically alter interpretations of genetic diversity. We propose a modified framework for calculating heterozygosity that reduces bias and improves the utility of heterozygosity as a measure of genetic diversity, whilst also highlighting the need for existing population genetic pipelines to be adjusted such that tri- and tetra-allelic sites be included in calculations.</p>","PeriodicalId":211,"journal":{"name":"Molecular Ecology Resources","volume":"24 4","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-03-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13947","citationCount":"0","resultStr":"{\"title\":\"Estimates of heterozygosity from single nucleotide polymorphism markers are context-dependent and often wrong\",\"authors\":\"Jarrod Sopniewski, Renee A. Catullo\",\"doi\":\"10.1111/1755-0998.13947\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Genetic diversity is frequently described using heterozygosity, particularly in a conservation context. Often, it is estimated using single nucleotide polymorphisms (SNPs); however, it has been shown that heterozygosity values calculated from SNPs can be biased by both study design and filtering parameters. Though solutions have been proposed to address these issues, our own work has found them to be inadequate in some circumstances. Here, we aimed to improve the reliability and comparability of heterozygosity estimates, specifically by investigating how sample size and missing data thresholds influenced the calculation of autosomal heterozygosity (heterozygosity calculated from across the genome, i.e. fixed and variable sites). We also explored how the standard practice of tri- and tetra-allelic site exclusion could bias heterozygosity estimates and influence eventual conclusions relating to genetic diversity. Across three distinct taxa (a frog, <i>Litoria rubella</i>; a tree, <i>Eucalyptus microcarpa</i>; and a grasshopper, <i>Keyacris scurra</i>), we found heterozygosity estimates to be meaningfully affected by sample size and missing data thresholds, partly due to the exclusion of tri- and tetra-allelic sites. These biases were inconsistent both between species and populations, with more diverse populations tending to have their estimates more severely affected, thus having potential to dramatically alter interpretations of genetic diversity. We propose a modified framework for calculating heterozygosity that reduces bias and improves the utility of heterozygosity as a measure of genetic diversity, whilst also highlighting the need for existing population genetic pipelines to be adjusted such that tri- and tetra-allelic sites be included in calculations.</p>\",\"PeriodicalId\":211,\"journal\":{\"name\":\"Molecular Ecology Resources\",\"volume\":\"24 4\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-03-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1755-0998.13947\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Ecology Resources\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13947\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Ecology Resources","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13947","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Estimates of heterozygosity from single nucleotide polymorphism markers are context-dependent and often wrong
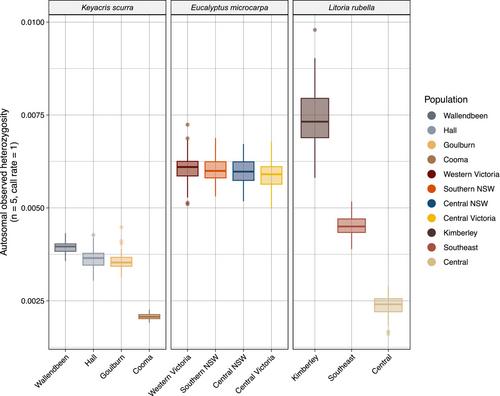
Genetic diversity is frequently described using heterozygosity, particularly in a conservation context. Often, it is estimated using single nucleotide polymorphisms (SNPs); however, it has been shown that heterozygosity values calculated from SNPs can be biased by both study design and filtering parameters. Though solutions have been proposed to address these issues, our own work has found them to be inadequate in some circumstances. Here, we aimed to improve the reliability and comparability of heterozygosity estimates, specifically by investigating how sample size and missing data thresholds influenced the calculation of autosomal heterozygosity (heterozygosity calculated from across the genome, i.e. fixed and variable sites). We also explored how the standard practice of tri- and tetra-allelic site exclusion could bias heterozygosity estimates and influence eventual conclusions relating to genetic diversity. Across three distinct taxa (a frog, Litoria rubella; a tree, Eucalyptus microcarpa; and a grasshopper, Keyacris scurra), we found heterozygosity estimates to be meaningfully affected by sample size and missing data thresholds, partly due to the exclusion of tri- and tetra-allelic sites. These biases were inconsistent both between species and populations, with more diverse populations tending to have their estimates more severely affected, thus having potential to dramatically alter interpretations of genetic diversity. We propose a modified framework for calculating heterozygosity that reduces bias and improves the utility of heterozygosity as a measure of genetic diversity, whilst also highlighting the need for existing population genetic pipelines to be adjusted such that tri- and tetra-allelic sites be included in calculations.
期刊介绍:
Molecular Ecology Resources promotes the creation of comprehensive resources for the scientific community, encompassing computer programs, statistical and molecular advancements, and a diverse array of molecular tools. Serving as a conduit for disseminating these resources, the journal targets a broad audience of researchers in the fields of evolution, ecology, and conservation. Articles in Molecular Ecology Resources are crafted to support investigations tackling significant questions within these disciplines.
In addition to original resource articles, Molecular Ecology Resources features Reviews, Opinions, and Comments relevant to the field. The journal also periodically releases Special Issues focusing on resource development within specific areas.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


