Fengyu Che, Chenhao Li, Liyu Zhang, Chenxi Qian, Lidangzhi Mo, Benchang Li, Haibin Wu, Lifang Wang, Ying Yang
{"title":"一个中国家庭中与言语和语言功能障碍相关的新型 FOXP2 变异及文献综述。","authors":"Fengyu Che, Chenhao Li, Liyu Zhang, Chenxi Qian, Lidangzhi Mo, Benchang Li, Haibin Wu, Lifang Wang, Ying Yang","doi":"10.1007/s13353-024-00849-0","DOIUrl":null,"url":null,"abstract":"<p><p>Since its initial identification, the Forkhead Box P2 gene (FOXP2) has maintained its singular status as the archetypal monogenic determinant implicated in Mendelian forms of human speech and language impairments. Despite the passage of two decades subsequent to its discovery, extant literature remains disproportionately sparse with regard to case-specific instances and loci of mutational perturbations. The objective of the current investigation centers on furnishing an enriched delineation of both its clinical manifestations and its mutational heterogeneity. Clinical phenotypes and peripheral blood samples were assiduously amassed from familial subjects. Whole-exome sequencing and Sanger sequencing methodologies were deployed for the unambiguous identification of potential genetic variants and for corroborating their co-segregation within the family pedigree. An exhaustive review of published literature focusing on patients manifesting speech and language disorders consequent to FOXP2 genetic anomalies was also undertaken. The investigation yielded the identification of a novel heterozygous variant, c.661del (p.L221Ffs*41), localized within the FOXP2 gene in the proband, an inheritance from his symptomatic mother. The proband presented with an array of symptoms, encompassing dysarthric speech, deficits in instruction comprehension, and communicative impediments. In comparison, the mother exhibited attenuated symptoms, including rudimentary verbalization capabilities punctuated by pronounced stuttering and dysarthria. A comprehensive analysis of articles archived in the Human Gene Mutation Database (HGMD) classified under \"DM\" disclosed the existence of 74 patients inclusive of the subjects under current examination, sub-divided into 19 patients with null variants, 5 patients with missense variants, and 50 patients with gross deletions or complex genomic rearrangements. A conspicuous predominance of delayed speech, impoverished current verbal abilities, verbal comprehension deficits, and learning difficulties were observed in patients harboring null or missense FOXP2 variants, as compared to their counterparts with gross deletions or complex rearrangements. Developmental delays, hypotonia, and craniofacial aberrations were exclusive to the latter cohort. The elucidated findings augment the existing corpus of knowledge on the genetic architecture influencing both the proband and his mother within this specified familial context. Of critical importance, these discoveries furnish a robust molecular framework conducive to the prenatal diagnostic evaluations of prospective progeny within this familial lineage.</p>","PeriodicalId":14891,"journal":{"name":"Journal of Applied Genetics","volume":" ","pages":"367-373"},"PeriodicalIF":2.0000,"publicationDate":"2024-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Novel FOXP2 variant associated with speech and language dysfunction in a Chinese family and literature review.\",\"authors\":\"Fengyu Che, Chenhao Li, Liyu Zhang, Chenxi Qian, Lidangzhi Mo, Benchang Li, Haibin Wu, Lifang Wang, Ying Yang\",\"doi\":\"10.1007/s13353-024-00849-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Since its initial identification, the Forkhead Box P2 gene (FOXP2) has maintained its singular status as the archetypal monogenic determinant implicated in Mendelian forms of human speech and language impairments. Despite the passage of two decades subsequent to its discovery, extant literature remains disproportionately sparse with regard to case-specific instances and loci of mutational perturbations. The objective of the current investigation centers on furnishing an enriched delineation of both its clinical manifestations and its mutational heterogeneity. Clinical phenotypes and peripheral blood samples were assiduously amassed from familial subjects. Whole-exome sequencing and Sanger sequencing methodologies were deployed for the unambiguous identification of potential genetic variants and for corroborating their co-segregation within the family pedigree. An exhaustive review of published literature focusing on patients manifesting speech and language disorders consequent to FOXP2 genetic anomalies was also undertaken. The investigation yielded the identification of a novel heterozygous variant, c.661del (p.L221Ffs*41), localized within the FOXP2 gene in the proband, an inheritance from his symptomatic mother. The proband presented with an array of symptoms, encompassing dysarthric speech, deficits in instruction comprehension, and communicative impediments. In comparison, the mother exhibited attenuated symptoms, including rudimentary verbalization capabilities punctuated by pronounced stuttering and dysarthria. A comprehensive analysis of articles archived in the Human Gene Mutation Database (HGMD) classified under \\\"DM\\\" disclosed the existence of 74 patients inclusive of the subjects under current examination, sub-divided into 19 patients with null variants, 5 patients with missense variants, and 50 patients with gross deletions or complex genomic rearrangements. A conspicuous predominance of delayed speech, impoverished current verbal abilities, verbal comprehension deficits, and learning difficulties were observed in patients harboring null or missense FOXP2 variants, as compared to their counterparts with gross deletions or complex rearrangements. Developmental delays, hypotonia, and craniofacial aberrations were exclusive to the latter cohort. The elucidated findings augment the existing corpus of knowledge on the genetic architecture influencing both the proband and his mother within this specified familial context. Of critical importance, these discoveries furnish a robust molecular framework conducive to the prenatal diagnostic evaluations of prospective progeny within this familial lineage.</p>\",\"PeriodicalId\":14891,\"journal\":{\"name\":\"Journal of Applied Genetics\",\"volume\":\" \",\"pages\":\"367-373\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2024-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Applied Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s13353-024-00849-0\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/2/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Applied Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s13353-024-00849-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Novel FOXP2 variant associated with speech and language dysfunction in a Chinese family and literature review.
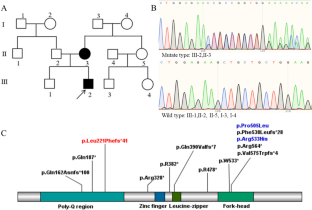
Since its initial identification, the Forkhead Box P2 gene (FOXP2) has maintained its singular status as the archetypal monogenic determinant implicated in Mendelian forms of human speech and language impairments. Despite the passage of two decades subsequent to its discovery, extant literature remains disproportionately sparse with regard to case-specific instances and loci of mutational perturbations. The objective of the current investigation centers on furnishing an enriched delineation of both its clinical manifestations and its mutational heterogeneity. Clinical phenotypes and peripheral blood samples were assiduously amassed from familial subjects. Whole-exome sequencing and Sanger sequencing methodologies were deployed for the unambiguous identification of potential genetic variants and for corroborating their co-segregation within the family pedigree. An exhaustive review of published literature focusing on patients manifesting speech and language disorders consequent to FOXP2 genetic anomalies was also undertaken. The investigation yielded the identification of a novel heterozygous variant, c.661del (p.L221Ffs*41), localized within the FOXP2 gene in the proband, an inheritance from his symptomatic mother. The proband presented with an array of symptoms, encompassing dysarthric speech, deficits in instruction comprehension, and communicative impediments. In comparison, the mother exhibited attenuated symptoms, including rudimentary verbalization capabilities punctuated by pronounced stuttering and dysarthria. A comprehensive analysis of articles archived in the Human Gene Mutation Database (HGMD) classified under "DM" disclosed the existence of 74 patients inclusive of the subjects under current examination, sub-divided into 19 patients with null variants, 5 patients with missense variants, and 50 patients with gross deletions or complex genomic rearrangements. A conspicuous predominance of delayed speech, impoverished current verbal abilities, verbal comprehension deficits, and learning difficulties were observed in patients harboring null or missense FOXP2 variants, as compared to their counterparts with gross deletions or complex rearrangements. Developmental delays, hypotonia, and craniofacial aberrations were exclusive to the latter cohort. The elucidated findings augment the existing corpus of knowledge on the genetic architecture influencing both the proband and his mother within this specified familial context. Of critical importance, these discoveries furnish a robust molecular framework conducive to the prenatal diagnostic evaluations of prospective progeny within this familial lineage.
期刊介绍:
The Journal of Applied Genetics is an international journal on genetics and genomics. It publishes peer-reviewed original papers, short communications (including case reports) and review articles focused on the research of applicative aspects of plant, human, animal and microbial genetics and genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


