Andrin Fluetsch, Elena Di Lascio, Grégori Gerebtzoff and Raquel Rodríguez-Pérez*,
{"title":"使深度学习 QSPR 模型适应特定的药物发现项目。","authors":"Andrin Fluetsch, Elena Di Lascio, Grégori Gerebtzoff and Raquel Rodríguez-Pérez*, ","doi":"10.1021/acs.molpharmaceut.3c01124","DOIUrl":null,"url":null,"abstract":"<p >Medicinal chemistry and drug design efforts can be assisted by machine learning (ML) models that relate the molecular structure to compound properties. Such quantitative structure–property relationship models are generally trained on large data sets that include diverse chemical series (global models). In the pharmaceutical industry, these ML global models are available across discovery projects as an “out-of-the-box” solution to assist in drug design, synthesis prioritization, and experiment selection. However, drug discovery projects typically focus on confined parts of the chemical space (e.g., chemical series), where global models might not be applicable. Local ML models are sometimes generated to focus on specific projects or series. Herein, ML-based global models, local models, and hybrid global-local strategies were benchmarked. Analyses were done for more than 300 drug discovery projects at Novartis and ten absorption, distribution, metabolism, and excretion (ADME) assays. In this work, hybrid global-local strategies based on transfer learning approaches were proposed to leverage both historical ADME data (global) and project-specific data (local) to adapt model predictions. Fine-tuning a pretrained global ML model (used for weights’ initialization, WI) was the top-performing method. Average improvements of mean absolute errors across all assays were 16% and 27% compared with global and local models, respectively. Interestingly, when the effect of training set size was analyzed, WI fine-tuning was found to be successful even in low-data scenarios (e.g., ∼10 molecules per project). Taken together, this work highlights the potential of domain adaptation in the field of molecular property predictions to refine existing pretrained models on a new compound data distribution.</p>","PeriodicalId":52,"journal":{"name":"Molecular Pharmaceutics","volume":"21 4","pages":"1817–1826"},"PeriodicalIF":4.5000,"publicationDate":"2024-02-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Adapting Deep Learning QSPR Models to Specific Drug Discovery Projects\",\"authors\":\"Andrin Fluetsch, Elena Di Lascio, Grégori Gerebtzoff and Raquel Rodríguez-Pérez*, \",\"doi\":\"10.1021/acs.molpharmaceut.3c01124\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Medicinal chemistry and drug design efforts can be assisted by machine learning (ML) models that relate the molecular structure to compound properties. Such quantitative structure–property relationship models are generally trained on large data sets that include diverse chemical series (global models). In the pharmaceutical industry, these ML global models are available across discovery projects as an “out-of-the-box” solution to assist in drug design, synthesis prioritization, and experiment selection. However, drug discovery projects typically focus on confined parts of the chemical space (e.g., chemical series), where global models might not be applicable. Local ML models are sometimes generated to focus on specific projects or series. Herein, ML-based global models, local models, and hybrid global-local strategies were benchmarked. Analyses were done for more than 300 drug discovery projects at Novartis and ten absorption, distribution, metabolism, and excretion (ADME) assays. In this work, hybrid global-local strategies based on transfer learning approaches were proposed to leverage both historical ADME data (global) and project-specific data (local) to adapt model predictions. Fine-tuning a pretrained global ML model (used for weights’ initialization, WI) was the top-performing method. Average improvements of mean absolute errors across all assays were 16% and 27% compared with global and local models, respectively. Interestingly, when the effect of training set size was analyzed, WI fine-tuning was found to be successful even in low-data scenarios (e.g., ∼10 molecules per project). Taken together, this work highlights the potential of domain adaptation in the field of molecular property predictions to refine existing pretrained models on a new compound data distribution.</p>\",\"PeriodicalId\":52,\"journal\":{\"name\":\"Molecular Pharmaceutics\",\"volume\":\"21 4\",\"pages\":\"1817–1826\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-02-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Pharmaceutics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.3c01124\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pharmaceutics","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.3c01124","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
摘要
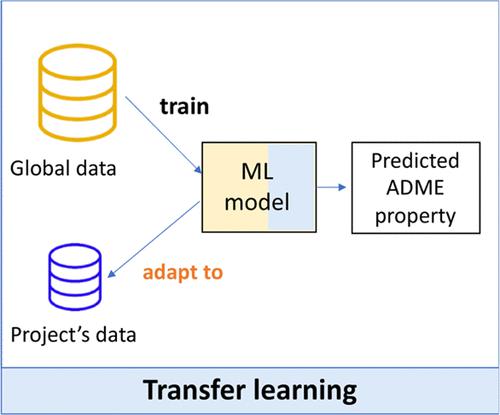
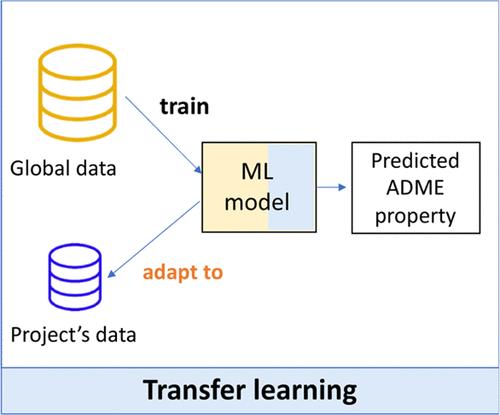
将分子结构与化合物性质联系起来的机器学习(ML)模型可以帮助药物化学和药物设计工作。此类定量结构-性质关系模型通常是在包含不同化学系列的大型数据集(全局模型)上训练出来的。在制药行业,这些 ML 全局模型作为 "开箱即用 "的解决方案,可用于各种发现项目,以协助药物设计、合成优先级排序和实验选择。然而,药物发现项目通常侧重于化学空间的有限部分(如化学系列),全局模型可能并不适用。有时会生成局部 ML 模型来关注特定项目或系列。在此,对基于 ML 的全局模型、局部模型和全局-局部混合策略进行了基准测试。我们对诺华公司的 300 多个药物发现项目和 10 项吸收、分布、代谢和排泄(ADME)检测进行了分析。在这项工作中,提出了基于迁移学习方法的全局-局部混合策略,以利用历史 ADME 数据(全局)和特定项目数据(局部)来调整模型预测。微调预训练的全局 ML 模型(用于权重初始化,WI)是表现最好的方法。与全局模型和局部模型相比,所有检测的平均绝对误差分别提高了 16% 和 27%。有趣的是,在分析训练集大小的影响时发现,即使在低数据量的情况下(例如每个项目只有 10 个分子),WI 微调也能取得成功。综上所述,这项工作凸显了领域适应在分子性质预测领域的潜力,可以在新的化合物数据分布上完善现有的预训练模型。
Adapting Deep Learning QSPR Models to Specific Drug Discovery Projects
Medicinal chemistry and drug design efforts can be assisted by machine learning (ML) models that relate the molecular structure to compound properties. Such quantitative structure–property relationship models are generally trained on large data sets that include diverse chemical series (global models). In the pharmaceutical industry, these ML global models are available across discovery projects as an “out-of-the-box” solution to assist in drug design, synthesis prioritization, and experiment selection. However, drug discovery projects typically focus on confined parts of the chemical space (e.g., chemical series), where global models might not be applicable. Local ML models are sometimes generated to focus on specific projects or series. Herein, ML-based global models, local models, and hybrid global-local strategies were benchmarked. Analyses were done for more than 300 drug discovery projects at Novartis and ten absorption, distribution, metabolism, and excretion (ADME) assays. In this work, hybrid global-local strategies based on transfer learning approaches were proposed to leverage both historical ADME data (global) and project-specific data (local) to adapt model predictions. Fine-tuning a pretrained global ML model (used for weights’ initialization, WI) was the top-performing method. Average improvements of mean absolute errors across all assays were 16% and 27% compared with global and local models, respectively. Interestingly, when the effect of training set size was analyzed, WI fine-tuning was found to be successful even in low-data scenarios (e.g., ∼10 molecules per project). Taken together, this work highlights the potential of domain adaptation in the field of molecular property predictions to refine existing pretrained models on a new compound data distribution.
期刊介绍:
Molecular Pharmaceutics publishes the results of original research that contributes significantly to the molecular mechanistic understanding of drug delivery and drug delivery systems. The journal encourages contributions describing research at the interface of drug discovery and drug development.
Scientific areas within the scope of the journal include physical and pharmaceutical chemistry, biochemistry and biophysics, molecular and cellular biology, and polymer and materials science as they relate to drug and drug delivery system efficacy. Mechanistic Drug Delivery and Drug Targeting research on modulating activity and efficacy of a drug or drug product is within the scope of Molecular Pharmaceutics. Theoretical and experimental peer-reviewed research articles, communications, reviews, and perspectives are welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


