{"title":"DFT 计算和 MO 理论提出的取代基定向效应的量子化学解释和 APT 原子电荷指数","authors":"Hongchang Shi","doi":"10.1002/poc.4580","DOIUrl":null,"url":null,"abstract":"<p>In organic chemistry textbooks, the electron resonance states are used to explain the orienting effect of benzene substituent. However, these resonance states do not exist in the C<sub>6</sub>H<sub>5</sub>-S + E<sup>+</sup> electrophilic addition, so the resonance state discussions actually are meaningless. The density functional theory (DFT) calculation shows that the C<sub>6</sub>H<sub>5</sub>-S + E<sup>+</sup> addition follows the transition state theory and needs to overcome a barrier. Benzene molecule C<sub>6</sub>H<sub>6</sub> has two degenerate highest occupied molecular orbitals (HOMOs), but after becoming substituted benzenes, the two HOMOs split to HOMO and HOMO-1 with different orbital energies and electron distributions. It makes the addition barrier on two or three benzene ring carbons is low, and the addition rate is fast, but the barrier of other carbons is high, and the rate is slow. Therefore, substituent splits the two degenerate HOMOs of benzene molecule, which is the origin of substituent orienting effect. For the substituted benzenes with halogen group -X and electron-releasing group, their HOMO images alone can determine that the two types of substituents are the ortho- and para-orienting groups, but for the substituted benzenes with electron-withdrawing group, their HOMO alone cannot determine whether the substituent is a meta-orienting group. The study found that atomic polar tensor (APT) charges are a good judgment index of substituent orienting effect because based on its physic-chemical model, the charges represent the number of active electrons on the benzene ring carbons. The DFT calculation gives the APT charges of 19 mono-substituted and 12 di-substituted benzenes and some aromatic molecules. The APT charges can all correctly determine their orienting effects, showing that APT atomic charge from quantum theoretical calculation is a good substituent orienting index.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"37 6","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2024-02-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Quantum chemical explanation and APT atomic charge index of substituent orienting effect proposed by DFT calculation and MO Theory\",\"authors\":\"Hongchang Shi\",\"doi\":\"10.1002/poc.4580\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In organic chemistry textbooks, the electron resonance states are used to explain the orienting effect of benzene substituent. However, these resonance states do not exist in the C<sub>6</sub>H<sub>5</sub>-S + E<sup>+</sup> electrophilic addition, so the resonance state discussions actually are meaningless. The density functional theory (DFT) calculation shows that the C<sub>6</sub>H<sub>5</sub>-S + E<sup>+</sup> addition follows the transition state theory and needs to overcome a barrier. Benzene molecule C<sub>6</sub>H<sub>6</sub> has two degenerate highest occupied molecular orbitals (HOMOs), but after becoming substituted benzenes, the two HOMOs split to HOMO and HOMO-1 with different orbital energies and electron distributions. It makes the addition barrier on two or three benzene ring carbons is low, and the addition rate is fast, but the barrier of other carbons is high, and the rate is slow. Therefore, substituent splits the two degenerate HOMOs of benzene molecule, which is the origin of substituent orienting effect. For the substituted benzenes with halogen group -X and electron-releasing group, their HOMO images alone can determine that the two types of substituents are the ortho- and para-orienting groups, but for the substituted benzenes with electron-withdrawing group, their HOMO alone cannot determine whether the substituent is a meta-orienting group. The study found that atomic polar tensor (APT) charges are a good judgment index of substituent orienting effect because based on its physic-chemical model, the charges represent the number of active electrons on the benzene ring carbons. The DFT calculation gives the APT charges of 19 mono-substituted and 12 di-substituted benzenes and some aromatic molecules. The APT charges can all correctly determine their orienting effects, showing that APT atomic charge from quantum theoretical calculation is a good substituent orienting index.</p>\",\"PeriodicalId\":16829,\"journal\":{\"name\":\"Journal of Physical Organic Chemistry\",\"volume\":\"37 6\",\"pages\":\"\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2024-02-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physical Organic Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/poc.4580\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4580","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
Quantum chemical explanation and APT atomic charge index of substituent orienting effect proposed by DFT calculation and MO Theory
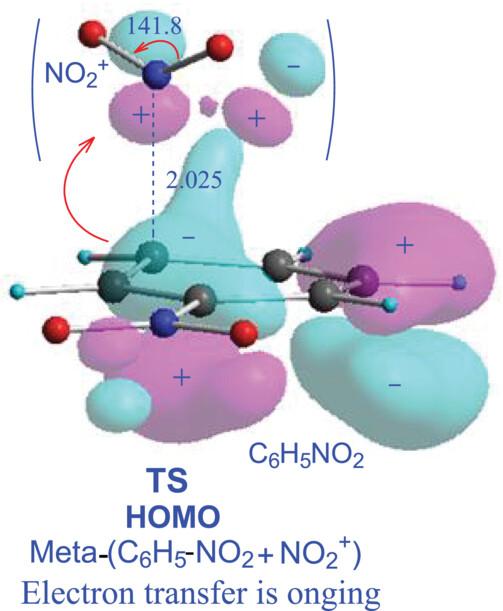
In organic chemistry textbooks, the electron resonance states are used to explain the orienting effect of benzene substituent. However, these resonance states do not exist in the C6H5-S + E+ electrophilic addition, so the resonance state discussions actually are meaningless. The density functional theory (DFT) calculation shows that the C6H5-S + E+ addition follows the transition state theory and needs to overcome a barrier. Benzene molecule C6H6 has two degenerate highest occupied molecular orbitals (HOMOs), but after becoming substituted benzenes, the two HOMOs split to HOMO and HOMO-1 with different orbital energies and electron distributions. It makes the addition barrier on two or three benzene ring carbons is low, and the addition rate is fast, but the barrier of other carbons is high, and the rate is slow. Therefore, substituent splits the two degenerate HOMOs of benzene molecule, which is the origin of substituent orienting effect. For the substituted benzenes with halogen group -X and electron-releasing group, their HOMO images alone can determine that the two types of substituents are the ortho- and para-orienting groups, but for the substituted benzenes with electron-withdrawing group, their HOMO alone cannot determine whether the substituent is a meta-orienting group. The study found that atomic polar tensor (APT) charges are a good judgment index of substituent orienting effect because based on its physic-chemical model, the charges represent the number of active electrons on the benzene ring carbons. The DFT calculation gives the APT charges of 19 mono-substituted and 12 di-substituted benzenes and some aromatic molecules. The APT charges can all correctly determine their orienting effects, showing that APT atomic charge from quantum theoretical calculation is a good substituent orienting index.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


