{"title":"利用一维和二维结构特征预测大数据集的晶体密度","authors":"Xianlan Li, Dingling Kong, Yue Luan, Lili Guo, Yanhua Lu, Wei Li, Meng Tang, Qingyou Zhang, Aimin Pang","doi":"10.1007/s11224-024-02279-4","DOIUrl":null,"url":null,"abstract":"<div><p>A large data set of over 30 thousand organic compounds containing carbon, nitrogen, oxygen, fluorine, and hydrogen was collected, and the density of each compound was predicted by 1D descriptors derived from its molecular formula and 2D descriptors derived from its constitutional structural features. The 2D structural features are composed of Benson’s groups, corrected groups, and 2D structural features of the whole molecular structures. All the descriptors were extracted by an in-house program in Java with a function to ensure that each atom (or bond) of molecules is represented by Benson’s groups once for atom-based (or bond-based) descriptors. Partial least square (PLS) and random forest (RF) methods were used separately to build models to predict the density. Further, the variable selection of descriptors was performed by variable importance of RF. For partial least square, the combination of the models constructed by descriptors based on the atoms and the bonds achieved the best results in this paper: for the cross-validation of the training set, the Pearson correlation coefficient (<i>R</i>) = 0.9270, mean absolute error (<i>MAE</i>) = 0.0270 g·cm<sup>−3</sup>, and root mean squared error (<i>RMSE</i>) = 0.0426 g·cm<sup>−3</sup>; for the prediction of the test set, <i>R</i> = 0.9454, <i>MAE</i> = 0.0263 g·cm<sup>−3</sup>, and <i>RMSE</i> = 0.0375 g·cm<sup>−3</sup>.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 5","pages":"1375 - 1385"},"PeriodicalIF":2.2000,"publicationDate":"2024-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The prediction of crystal densities of a big data set using 1D and 2D structure features\",\"authors\":\"Xianlan Li, Dingling Kong, Yue Luan, Lili Guo, Yanhua Lu, Wei Li, Meng Tang, Qingyou Zhang, Aimin Pang\",\"doi\":\"10.1007/s11224-024-02279-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>A large data set of over 30 thousand organic compounds containing carbon, nitrogen, oxygen, fluorine, and hydrogen was collected, and the density of each compound was predicted by 1D descriptors derived from its molecular formula and 2D descriptors derived from its constitutional structural features. The 2D structural features are composed of Benson’s groups, corrected groups, and 2D structural features of the whole molecular structures. All the descriptors were extracted by an in-house program in Java with a function to ensure that each atom (or bond) of molecules is represented by Benson’s groups once for atom-based (or bond-based) descriptors. Partial least square (PLS) and random forest (RF) methods were used separately to build models to predict the density. Further, the variable selection of descriptors was performed by variable importance of RF. For partial least square, the combination of the models constructed by descriptors based on the atoms and the bonds achieved the best results in this paper: for the cross-validation of the training set, the Pearson correlation coefficient (<i>R</i>) = 0.9270, mean absolute error (<i>MAE</i>) = 0.0270 g·cm<sup>−3</sup>, and root mean squared error (<i>RMSE</i>) = 0.0426 g·cm<sup>−3</sup>; for the prediction of the test set, <i>R</i> = 0.9454, <i>MAE</i> = 0.0263 g·cm<sup>−3</sup>, and <i>RMSE</i> = 0.0375 g·cm<sup>−3</sup>.</p></div>\",\"PeriodicalId\":780,\"journal\":{\"name\":\"Structural Chemistry\",\"volume\":\"35 5\",\"pages\":\"1375 - 1385\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-02-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Structural Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s11224-024-02279-4\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02279-4","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
The prediction of crystal densities of a big data set using 1D and 2D structure features
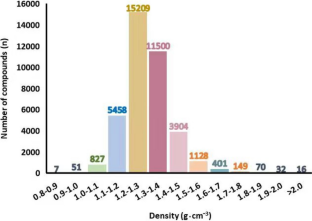
A large data set of over 30 thousand organic compounds containing carbon, nitrogen, oxygen, fluorine, and hydrogen was collected, and the density of each compound was predicted by 1D descriptors derived from its molecular formula and 2D descriptors derived from its constitutional structural features. The 2D structural features are composed of Benson’s groups, corrected groups, and 2D structural features of the whole molecular structures. All the descriptors were extracted by an in-house program in Java with a function to ensure that each atom (or bond) of molecules is represented by Benson’s groups once for atom-based (or bond-based) descriptors. Partial least square (PLS) and random forest (RF) methods were used separately to build models to predict the density. Further, the variable selection of descriptors was performed by variable importance of RF. For partial least square, the combination of the models constructed by descriptors based on the atoms and the bonds achieved the best results in this paper: for the cross-validation of the training set, the Pearson correlation coefficient (R) = 0.9270, mean absolute error (MAE) = 0.0270 g·cm−3, and root mean squared error (RMSE) = 0.0426 g·cm−3; for the prediction of the test set, R = 0.9454, MAE = 0.0263 g·cm−3, and RMSE = 0.0375 g·cm−3.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


