Lingling Wang, Qianqian Zhang, Henry H. Y. Tong, Xiaojun Yao, Huanxiang Liu, Guohui Li
{"title":"揭开钾通道秘密的计算方法:结构、机理和药物设计","authors":"Lingling Wang, Qianqian Zhang, Henry H. Y. Tong, Xiaojun Yao, Huanxiang Liu, Guohui Li","doi":"10.1002/wcms.1704","DOIUrl":null,"url":null,"abstract":"<p>Potassium (K<sup>+</sup>) channels play vital roles in various physiological functions, including regulating K<sup>+</sup> flow in cell membranes, impacting nervous system signal transduction, neuronal firing, muscle contraction, neurotransmitters, and enzyme secretion. Their activation and switch-off are directly linked to diseases like arrhythmias, atrial fibrillation, and pain etc. Although the experimental methods play important roles in the studying the structure and function of K<sup>+</sup> channels, they are still some limitations to enclose the dynamic molecular processes and the corresponding mechanisms of conformational changes during ion transport, permeation, and gating control. Relatively, computational methods have obvious advantages in studying such problems compared with experimental methods. Recently, more and more three-dimensional structures of K<sup>+</sup> channels have been disclosed based on experimental methods and in silico prediction methods, which provide a good chance to study the molecular mechanism of conformational changes related to the functional regulations of K<sup>+</sup> channels. Based on these structural details, molecular dynamics simulations together with related methods such as enhanced sampling and free energy calculations, have been widely used to reveal the conformational dynamics, ion conductance, ion channel gating, and ligand binding mechanisms. Additionally, the accessibility of structures also provides a large space for structure-based drug design. This review mainly addresses the recent progress of computational methods in the structure, mechanism, and drug design of K<sup>+</sup> channels. After summarizing the progress in these fields, we also give our opinion on the future direction in the area of K<sup>+</sup> channel research combined with the cutting edge of computational methods.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":16.8000,"publicationDate":"2024-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational methods for unlocking the secrets of potassium channels: Structure, mechanism, and drug design\",\"authors\":\"Lingling Wang, Qianqian Zhang, Henry H. Y. Tong, Xiaojun Yao, Huanxiang Liu, Guohui Li\",\"doi\":\"10.1002/wcms.1704\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Potassium (K<sup>+</sup>) channels play vital roles in various physiological functions, including regulating K<sup>+</sup> flow in cell membranes, impacting nervous system signal transduction, neuronal firing, muscle contraction, neurotransmitters, and enzyme secretion. Their activation and switch-off are directly linked to diseases like arrhythmias, atrial fibrillation, and pain etc. Although the experimental methods play important roles in the studying the structure and function of K<sup>+</sup> channels, they are still some limitations to enclose the dynamic molecular processes and the corresponding mechanisms of conformational changes during ion transport, permeation, and gating control. Relatively, computational methods have obvious advantages in studying such problems compared with experimental methods. Recently, more and more three-dimensional structures of K<sup>+</sup> channels have been disclosed based on experimental methods and in silico prediction methods, which provide a good chance to study the molecular mechanism of conformational changes related to the functional regulations of K<sup>+</sup> channels. Based on these structural details, molecular dynamics simulations together with related methods such as enhanced sampling and free energy calculations, have been widely used to reveal the conformational dynamics, ion conductance, ion channel gating, and ligand binding mechanisms. Additionally, the accessibility of structures also provides a large space for structure-based drug design. This review mainly addresses the recent progress of computational methods in the structure, mechanism, and drug design of K<sup>+</sup> channels. After summarizing the progress in these fields, we also give our opinion on the future direction in the area of K<sup>+</sup> channel research combined with the cutting edge of computational methods.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":16.8000,\"publicationDate\":\"2024-02-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1704\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1704","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Computational methods for unlocking the secrets of potassium channels: Structure, mechanism, and drug design
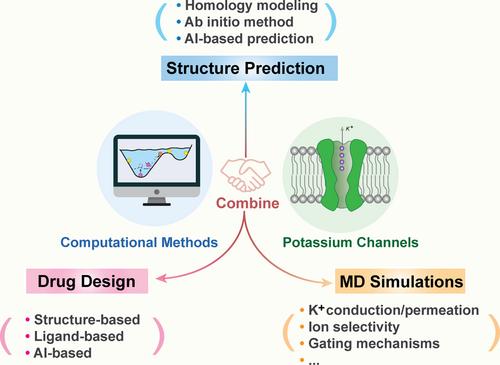
Potassium (K+) channels play vital roles in various physiological functions, including regulating K+ flow in cell membranes, impacting nervous system signal transduction, neuronal firing, muscle contraction, neurotransmitters, and enzyme secretion. Their activation and switch-off are directly linked to diseases like arrhythmias, atrial fibrillation, and pain etc. Although the experimental methods play important roles in the studying the structure and function of K+ channels, they are still some limitations to enclose the dynamic molecular processes and the corresponding mechanisms of conformational changes during ion transport, permeation, and gating control. Relatively, computational methods have obvious advantages in studying such problems compared with experimental methods. Recently, more and more three-dimensional structures of K+ channels have been disclosed based on experimental methods and in silico prediction methods, which provide a good chance to study the molecular mechanism of conformational changes related to the functional regulations of K+ channels. Based on these structural details, molecular dynamics simulations together with related methods such as enhanced sampling and free energy calculations, have been widely used to reveal the conformational dynamics, ion conductance, ion channel gating, and ligand binding mechanisms. Additionally, the accessibility of structures also provides a large space for structure-based drug design. This review mainly addresses the recent progress of computational methods in the structure, mechanism, and drug design of K+ channels. After summarizing the progress in these fields, we also give our opinion on the future direction in the area of K+ channel research combined with the cutting edge of computational methods.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


