{"title":"沉默 METTL3 能以 YTHDF2 的方式抑制心肌细胞因 SLC7A11 的 m6A 修饰而发生的铁变态反应。","authors":"Zengyao Tang, Xin Huang, Hanying Mei, Zeqi Zheng","doi":"10.1007/s10863-024-10006-1","DOIUrl":null,"url":null,"abstract":"<p><p>Myocardial infarction (MI) is the main cause of heart failure (HF). N6-methyladenosine (m6A) methylation is associated with the progression of HF. The study aimed to explore whether METTL3 regulates ferroptosis of cardiomyocytes in HF. We evaluated ferroptosis by detecting lactic dehydrogenase (LDH) release, lipid reactive oxygen species (ROS), Fe<sup>2+</sup>, glutathione (GSH), and malonaldehyde (MDA) levels. M6A methylation was assessed using methylated RNA immunoprecipitation assay. The binding relationship was assessed using RNA immunoprecipitation assays. The mRNA stability was assessed using actinomycin D treatment. The results showed that METTL3 was upregulated in oxygen glucose deprivation/recovery (OGD/R) cells, which knockdown suppressed OGD/R-induced ferroptosis. Moreover, METTL3 could bind to SLC7A11, promoting m6A methylation of SLC7A11. Silencing of SLC7A11 abrogated the suppression of ferroptosis induced by METTL3 knockdown. Additionally, YTHDF2 was the reader that recognized the methylation of SLC7A11, reducing the stability of SLC7A11. The silencing of METTL3 inhibited OGD/R-induced ferroptosis by suppressing the m6A methylation of SLC7A11, which is recognized by YTHDF2. The findings suggested that METTL3-mediated ferroptosis might be a new strategy for MI-induced HF therapy.</p>","PeriodicalId":15080,"journal":{"name":"Journal of Bioenergetics and Biomembranes","volume":" ","pages":"149-157"},"PeriodicalIF":3.0000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Silencing of METTL3 suppressed ferroptosis of myocardial cells by m6A modification of SLC7A11 in a YTHDF2 manner.\",\"authors\":\"Zengyao Tang, Xin Huang, Hanying Mei, Zeqi Zheng\",\"doi\":\"10.1007/s10863-024-10006-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Myocardial infarction (MI) is the main cause of heart failure (HF). N6-methyladenosine (m6A) methylation is associated with the progression of HF. The study aimed to explore whether METTL3 regulates ferroptosis of cardiomyocytes in HF. We evaluated ferroptosis by detecting lactic dehydrogenase (LDH) release, lipid reactive oxygen species (ROS), Fe<sup>2+</sup>, glutathione (GSH), and malonaldehyde (MDA) levels. M6A methylation was assessed using methylated RNA immunoprecipitation assay. The binding relationship was assessed using RNA immunoprecipitation assays. The mRNA stability was assessed using actinomycin D treatment. The results showed that METTL3 was upregulated in oxygen glucose deprivation/recovery (OGD/R) cells, which knockdown suppressed OGD/R-induced ferroptosis. Moreover, METTL3 could bind to SLC7A11, promoting m6A methylation of SLC7A11. Silencing of SLC7A11 abrogated the suppression of ferroptosis induced by METTL3 knockdown. Additionally, YTHDF2 was the reader that recognized the methylation of SLC7A11, reducing the stability of SLC7A11. The silencing of METTL3 inhibited OGD/R-induced ferroptosis by suppressing the m6A methylation of SLC7A11, which is recognized by YTHDF2. The findings suggested that METTL3-mediated ferroptosis might be a new strategy for MI-induced HF therapy.</p>\",\"PeriodicalId\":15080,\"journal\":{\"name\":\"Journal of Bioenergetics and Biomembranes\",\"volume\":\" \",\"pages\":\"149-157\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Bioenergetics and Biomembranes\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s10863-024-10006-1\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/2/6 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOPHYSICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Bioenergetics and Biomembranes","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s10863-024-10006-1","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/2/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOPHYSICS","Score":null,"Total":0}
Silencing of METTL3 suppressed ferroptosis of myocardial cells by m6A modification of SLC7A11 in a YTHDF2 manner.
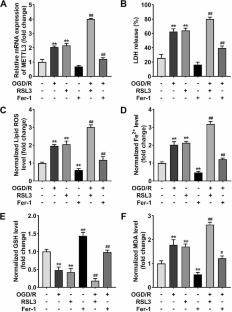
Myocardial infarction (MI) is the main cause of heart failure (HF). N6-methyladenosine (m6A) methylation is associated with the progression of HF. The study aimed to explore whether METTL3 regulates ferroptosis of cardiomyocytes in HF. We evaluated ferroptosis by detecting lactic dehydrogenase (LDH) release, lipid reactive oxygen species (ROS), Fe2+, glutathione (GSH), and malonaldehyde (MDA) levels. M6A methylation was assessed using methylated RNA immunoprecipitation assay. The binding relationship was assessed using RNA immunoprecipitation assays. The mRNA stability was assessed using actinomycin D treatment. The results showed that METTL3 was upregulated in oxygen glucose deprivation/recovery (OGD/R) cells, which knockdown suppressed OGD/R-induced ferroptosis. Moreover, METTL3 could bind to SLC7A11, promoting m6A methylation of SLC7A11. Silencing of SLC7A11 abrogated the suppression of ferroptosis induced by METTL3 knockdown. Additionally, YTHDF2 was the reader that recognized the methylation of SLC7A11, reducing the stability of SLC7A11. The silencing of METTL3 inhibited OGD/R-induced ferroptosis by suppressing the m6A methylation of SLC7A11, which is recognized by YTHDF2. The findings suggested that METTL3-mediated ferroptosis might be a new strategy for MI-induced HF therapy.
期刊介绍:
The Journal of Bioenergetics and Biomembranes is an international journal devoted to the publication of original research that contributes to fundamental knowledge in the areas of bioenergetics, biomembranes, and transport, including oxidative phosphorylation, photosynthesis, muscle contraction, as well as cellular and systemic metabolism. The timely research in this international journal benefits biophysicists, membrane biologists, cell biologists, biochemists, molecular biologists, physiologists, endocrinologists, and bio-organic chemists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


