{"title":"通过偏置交换自适应偏置分子动力学分析跨膜同源二聚体的自由能图谱","authors":"Shingo Ito , Yuji Sugita","doi":"10.1016/j.bpc.2024.107190","DOIUrl":null,"url":null,"abstract":"<div><p><span><span><span>Membrane proteins play essential roles in various biological functions within the cell. One of the most common functional regulations involves the dimerization of two single-pass transmembrane (TM) helices. </span>Glycophorin A (GpA) and </span>amyloid precursor protein (APP) form TM </span>homodimers<span> in the membrane, which have been investigated both experimentally and computationally. The homodimer structures are well characterized using only four collective variables (CVs) when each TM helix is stable. The CVs are the interhelical distance, the crossing angle, and the Crick angles for two TM helices. However, conformational sampling with multi-dimensional replica-exchange umbrella sampling<span><span> (REUS) requires too many replicas to sample all the CVs for exploring the conformational landscapes. Here, we show that the bias-exchange adaptively biased molecular dynamics<span> (BE-ABMD) with the four CVs effectively explores the free-energy landscapes of the TM helix dimers of GpA, wild-type APP and its mutants in the IMM1 implicit membrane. Compared to the original ABMD, the bias-exchange algorithm in BE-ABMD can provide a more rapidly converged conformational landscape. The BE-ABMD simulations could also reveal TM packing interfaces of the membrane proteins and the dependence of the free-energy landscapes on the membrane thickness. This approach is valuable for numerous other applications, including those involving explicit solvent and a </span></span>lipid bilayer<span> in all-atom force fields or Martini coarse-grained models, and enhances our understanding of protein-protein interactions in biological membranes.</span></span></span></p></div>","PeriodicalId":8979,"journal":{"name":"Biophysical chemistry","volume":"307 ","pages":"Article 107190"},"PeriodicalIF":3.3000,"publicationDate":"2024-01-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Free-energy landscapes of transmembrane homodimers by bias-exchange adaptively biased molecular dynamics\",\"authors\":\"Shingo Ito , Yuji Sugita\",\"doi\":\"10.1016/j.bpc.2024.107190\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><span><span><span>Membrane proteins play essential roles in various biological functions within the cell. One of the most common functional regulations involves the dimerization of two single-pass transmembrane (TM) helices. </span>Glycophorin A (GpA) and </span>amyloid precursor protein (APP) form TM </span>homodimers<span> in the membrane, which have been investigated both experimentally and computationally. The homodimer structures are well characterized using only four collective variables (CVs) when each TM helix is stable. The CVs are the interhelical distance, the crossing angle, and the Crick angles for two TM helices. However, conformational sampling with multi-dimensional replica-exchange umbrella sampling<span><span> (REUS) requires too many replicas to sample all the CVs for exploring the conformational landscapes. Here, we show that the bias-exchange adaptively biased molecular dynamics<span> (BE-ABMD) with the four CVs effectively explores the free-energy landscapes of the TM helix dimers of GpA, wild-type APP and its mutants in the IMM1 implicit membrane. Compared to the original ABMD, the bias-exchange algorithm in BE-ABMD can provide a more rapidly converged conformational landscape. The BE-ABMD simulations could also reveal TM packing interfaces of the membrane proteins and the dependence of the free-energy landscapes on the membrane thickness. This approach is valuable for numerous other applications, including those involving explicit solvent and a </span></span>lipid bilayer<span> in all-atom force fields or Martini coarse-grained models, and enhances our understanding of protein-protein interactions in biological membranes.</span></span></span></p></div>\",\"PeriodicalId\":8979,\"journal\":{\"name\":\"Biophysical chemistry\",\"volume\":\"307 \",\"pages\":\"Article 107190\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-01-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biophysical chemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S030146222400019X\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S030146222400019X","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Free-energy landscapes of transmembrane homodimers by bias-exchange adaptively biased molecular dynamics
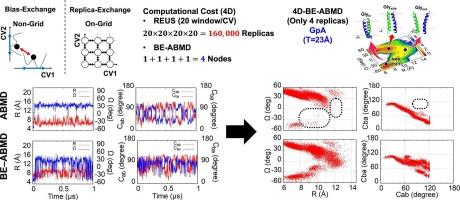
Membrane proteins play essential roles in various biological functions within the cell. One of the most common functional regulations involves the dimerization of two single-pass transmembrane (TM) helices. Glycophorin A (GpA) and amyloid precursor protein (APP) form TM homodimers in the membrane, which have been investigated both experimentally and computationally. The homodimer structures are well characterized using only four collective variables (CVs) when each TM helix is stable. The CVs are the interhelical distance, the crossing angle, and the Crick angles for two TM helices. However, conformational sampling with multi-dimensional replica-exchange umbrella sampling (REUS) requires too many replicas to sample all the CVs for exploring the conformational landscapes. Here, we show that the bias-exchange adaptively biased molecular dynamics (BE-ABMD) with the four CVs effectively explores the free-energy landscapes of the TM helix dimers of GpA, wild-type APP and its mutants in the IMM1 implicit membrane. Compared to the original ABMD, the bias-exchange algorithm in BE-ABMD can provide a more rapidly converged conformational landscape. The BE-ABMD simulations could also reveal TM packing interfaces of the membrane proteins and the dependence of the free-energy landscapes on the membrane thickness. This approach is valuable for numerous other applications, including those involving explicit solvent and a lipid bilayer in all-atom force fields or Martini coarse-grained models, and enhances our understanding of protein-protein interactions in biological membranes.
期刊介绍:
Biophysical Chemistry publishes original work and reviews in the areas of chemistry and physics directly impacting biological phenomena. Quantitative analysis of the properties of biological macromolecules, biologically active molecules, macromolecular assemblies and cell components in terms of kinetics, thermodynamics, spatio-temporal organization, NMR and X-ray structural biology, as well as single-molecule detection represent a major focus of the journal. Theoretical and computational treatments of biomacromolecular systems, macromolecular interactions, regulatory control and systems biology are also of interest to the journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


