Id2 通过破坏 Tcf3-LSD1 复合物的组装,从表观遗传学上控制 CD8+ T 细胞的衰竭。
IF 21.8
1区 医学
Q1 IMMUNOLOGY
引用次数: 0
摘要
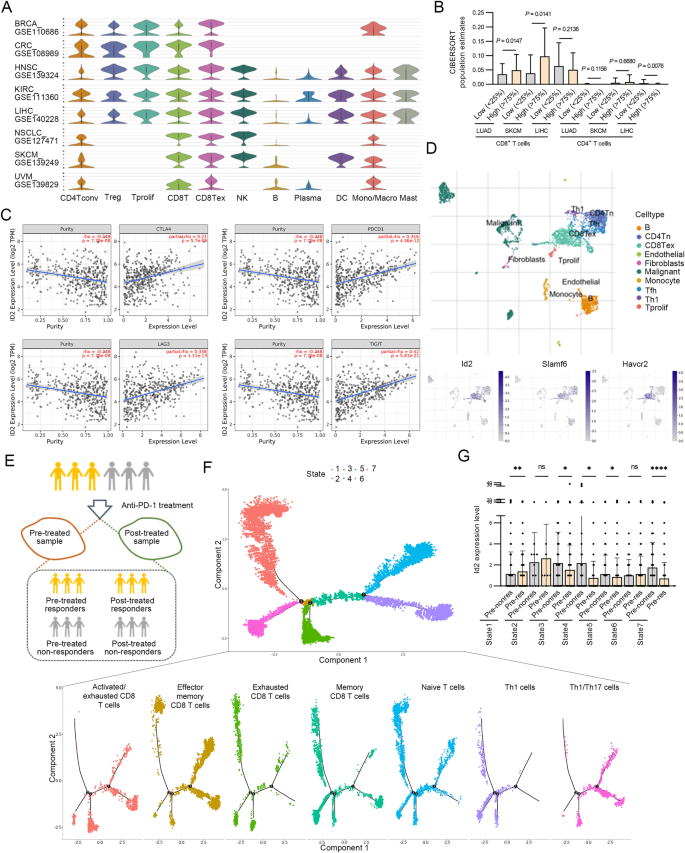
CD8+ T细胞衰竭是一种促进肿瘤进展的功能障碍状态,其特征是产生Slamf6+祖细胞衰竭(Texprog)和Tim-3+终末衰竭(Texterm)亚群。DNA 结合蛋白 2 抑制剂(Id2)已被证明在 T 细胞发育和 CD8+ T 细胞免疫中发挥重要作用。然而,Id2在CD8+ T细胞衰竭中的作用尚不清楚。在这里,我们发现Id2转录和表观遗传调控Texprog细胞的生成及其向Texterm细胞的转化。基因缺失Id2会抑制CD8+ T细胞介导的免疫反应和干样CD8+ T细胞亚群的维持,抑制PD-1阻断并增加肿瘤易感性。从机理上讲,Id2 通过其 HLH 结构域结合并破坏 Tcf3-Tal1 转录调控复合物的组装,从而通过阻止 Tcf3 与组蛋白赖氨酸去甲基化酶 LSD1 的相互作用来调节 Slamf6 启动子的染色质可及性。因此,Id2增加了Slamf6启动子中Tcf3占据的E-box上允许的H3K4me2标记的丰度,调节了Slamf6启动子的染色质可及性,并从表观遗传学上调控了Slamf6+ Texprog细胞的产生。LSD1抑制剂GSK2879552能挽救肿瘤小鼠的Id2基因敲除表型。抑制 LSD1 会增加肿瘤中 Slamf6+Tim-3- Texprog 细胞的数量,并提高 Id2 基因敲除的 CD8+ T 细胞中 Tcf1 的表达水平。这项研究表明,Id2介导的转录和表观遗传修饰推动了CD8+ T细胞的分层衰竭,所获得的机理认识可能对治疗干预肿瘤免疫逃避有意义。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Id2 epigenetically controls CD8+ T-cell exhaustion by disrupting the assembly of the Tcf3-LSD1 complex
CD8+ T-cell exhaustion is a state of dysfunction that promotes tumor progression and is marked by the generation of Slamf6+ progenitor exhausted (Texprog) and Tim-3+ terminally exhausted (Texterm) subpopulations. Inhibitor of DNA binding protein 2 (Id2) has been shown to play important roles in T-cell development and CD8+ T-cell immunity. However, the role of Id2 in CD8+ T-cell exhaustion is unclear. Here, we found that Id2 transcriptionally and epigenetically regulates the generation of Texprog cells and their conversion to Texterm cells. Genetic deletion of Id2 dampens CD8+ T-cell-mediated immune responses and the maintenance of stem-like CD8+ T-cell subpopulations, suppresses PD-1 blockade and increases tumor susceptibility. Mechanistically, through its HLH domain, Id2 binds and disrupts the assembly of the Tcf3-Tal1 transcriptional regulatory complex, and thus modulates chromatin accessibility at the Slamf6 promoter by preventing the interaction of Tcf3 with the histone lysine demethylase LSD1. Therefore, Id2 increases the abundance of the permissive H3K4me2 mark on the Tcf3-occupied E-boxes in the Slamf6 promoter, modulates chromatin accessibility at the Slamf6 promoter and epigenetically regulates the generation of Slamf6+ Texprog cells. An LSD1 inhibitor GSK2879552 can rescue the Id2 knockout phenotype in tumor-bearing mice. Inhibition of LSD1 increases the abundance of Slamf6+Tim-3− Texprog cells in tumors and the expression level of Tcf1 in Id2-deleted CD8+ T cells. This study demonstrates that Id2-mediated transcriptional and epigenetic modification drives hierarchical CD8+ T-cell exhaustion, and the mechanistic insights gained may have implications for therapeutic intervention with tumor immune evasion.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
31.20
自引率
1.20%
发文量
903
审稿时长
1 months
期刊介绍:
Cellular & Molecular Immunology, a monthly journal from the Chinese Society of Immunology and the University of Science and Technology of China, serves as a comprehensive platform covering both basic immunology research and clinical applications. The journal publishes a variety of article types, including Articles, Review Articles, Mini Reviews, and Short Communications, focusing on diverse aspects of cellular and molecular immunology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


