Hamid Aziz , Aamer Saeed , Jim Simpson , Tuncer Hökelek , Erum Jabeen
{"title":"1-(4-甲基苯甲酰基)硫脲的合成、单晶 X 射线结构测定、Hirshfeld 表面评价、空隙分析、相互作用能、能量框架和密度泛函理论研究","authors":"Hamid Aziz , Aamer Saeed , Jim Simpson , Tuncer Hökelek , Erum Jabeen","doi":"10.1016/j.cdc.2024.101121","DOIUrl":null,"url":null,"abstract":"<div><p>The current research work successfully presents facile synthesis, single crystal X-ray structure determination, Hirshfeld surface (HS) analysis, and Density functional theory (DFT) studies of 1-(4-methylbenzoyl)thiourea. Accordingly, the synthesized compound crystallized in triclinic <em>P -1</em> space group with <em>a</em> = 7.3464 (5) Å, <em>b</em> = 8.2696 (5) Å, <em>c</em> = 8.5337 (5) Å, α = 88.349 (5)°, β = 72.206 (5)°, γ = 69.901 (6)°, <em>Z</em> = 2 and <em>V</em> = 461.89 (5) Å<sup>3</sup>. In the crystal structure, the molecules are linked through intermolecular N—H···O, C—H···O, C—H···S and N—H···S bifurcated hydrogen bonds, enclosing R<sub>2</sub><sup>2</sup>(8), R<sub>2</sub><sup>2</sup>(14) and R<sub>4</sub><sup>4</sup>(11) ring motifs, into 2D network. HS analysis explored weak intermolecular interactions and indicated significant contributions responsible for crystal packing are H…H (41.0%), H…S/S…H (21.6%), H…C/C…H (12.7%) and H…O/O…H (9.9%) interactions, where hydrogen bonding and van der Waals contacts were dominant. Crystal voids analysis confirmed the absence of any large cavity within the packed crystal, suggesting higher mechanical stability of the crystal. Furthermore, analysıs of the electrostatic, dispersion and total energy frameworks augmented stabilization through electrostatic energy contribution. Moreover, optimized molecular structure, using DFT at B3LYP/6–311G(d,p) level was compared with the experimentally determined one. HOMO–LUMO energy gaps were determined and the molecular electrostatic potential (MESP) surfaces are calculated at B3LYP/6–31 G level to predict possible sites for electrophilic and nucleophilic attacks. DFT computations, through stable terminal HOMO and whole molecule LUMO spread, and significant polarity predicted amphoteric behavior of the synthesized 1-(4-methylbenzoyl)thiourea for its reaction with an electrophile or nucleophile.</p></div>","PeriodicalId":269,"journal":{"name":"Chemical Data Collections","volume":"50 ","pages":"Article 101121"},"PeriodicalIF":2.7000,"publicationDate":"2024-01-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Synthesis, single crystal X-ray structure determination, Hirshfeld surface evaluation, void analysis, interaction energies, energy frameworks and density functional theory studies of 1-(4-methylbenzoyl)thiourea\",\"authors\":\"Hamid Aziz , Aamer Saeed , Jim Simpson , Tuncer Hökelek , Erum Jabeen\",\"doi\":\"10.1016/j.cdc.2024.101121\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The current research work successfully presents facile synthesis, single crystal X-ray structure determination, Hirshfeld surface (HS) analysis, and Density functional theory (DFT) studies of 1-(4-methylbenzoyl)thiourea. Accordingly, the synthesized compound crystallized in triclinic <em>P -1</em> space group with <em>a</em> = 7.3464 (5) Å, <em>b</em> = 8.2696 (5) Å, <em>c</em> = 8.5337 (5) Å, α = 88.349 (5)°, β = 72.206 (5)°, γ = 69.901 (6)°, <em>Z</em> = 2 and <em>V</em> = 461.89 (5) Å<sup>3</sup>. In the crystal structure, the molecules are linked through intermolecular N—H···O, C—H···O, C—H···S and N—H···S bifurcated hydrogen bonds, enclosing R<sub>2</sub><sup>2</sup>(8), R<sub>2</sub><sup>2</sup>(14) and R<sub>4</sub><sup>4</sup>(11) ring motifs, into 2D network. HS analysis explored weak intermolecular interactions and indicated significant contributions responsible for crystal packing are H…H (41.0%), H…S/S…H (21.6%), H…C/C…H (12.7%) and H…O/O…H (9.9%) interactions, where hydrogen bonding and van der Waals contacts were dominant. Crystal voids analysis confirmed the absence of any large cavity within the packed crystal, suggesting higher mechanical stability of the crystal. Furthermore, analysıs of the electrostatic, dispersion and total energy frameworks augmented stabilization through electrostatic energy contribution. Moreover, optimized molecular structure, using DFT at B3LYP/6–311G(d,p) level was compared with the experimentally determined one. HOMO–LUMO energy gaps were determined and the molecular electrostatic potential (MESP) surfaces are calculated at B3LYP/6–31 G level to predict possible sites for electrophilic and nucleophilic attacks. DFT computations, through stable terminal HOMO and whole molecule LUMO spread, and significant polarity predicted amphoteric behavior of the synthesized 1-(4-methylbenzoyl)thiourea for its reaction with an electrophile or nucleophile.</p></div>\",\"PeriodicalId\":269,\"journal\":{\"name\":\"Chemical Data Collections\",\"volume\":\"50 \",\"pages\":\"Article 101121\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2024-01-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chemical Data Collections\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2405830024000090\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Data Collections","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2405830024000090","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
Synthesis, single crystal X-ray structure determination, Hirshfeld surface evaluation, void analysis, interaction energies, energy frameworks and density functional theory studies of 1-(4-methylbenzoyl)thiourea
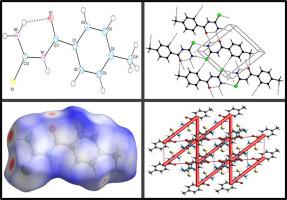
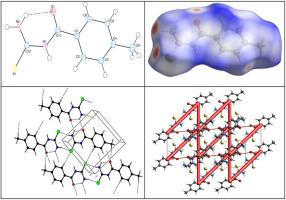
The current research work successfully presents facile synthesis, single crystal X-ray structure determination, Hirshfeld surface (HS) analysis, and Density functional theory (DFT) studies of 1-(4-methylbenzoyl)thiourea. Accordingly, the synthesized compound crystallized in triclinic P -1 space group with a = 7.3464 (5) Å, b = 8.2696 (5) Å, c = 8.5337 (5) Å, α = 88.349 (5)°, β = 72.206 (5)°, γ = 69.901 (6)°, Z = 2 and V = 461.89 (5) Å3. In the crystal structure, the molecules are linked through intermolecular N—H···O, C—H···O, C—H···S and N—H···S bifurcated hydrogen bonds, enclosing R22(8), R22(14) and R44(11) ring motifs, into 2D network. HS analysis explored weak intermolecular interactions and indicated significant contributions responsible for crystal packing are H…H (41.0%), H…S/S…H (21.6%), H…C/C…H (12.7%) and H…O/O…H (9.9%) interactions, where hydrogen bonding and van der Waals contacts were dominant. Crystal voids analysis confirmed the absence of any large cavity within the packed crystal, suggesting higher mechanical stability of the crystal. Furthermore, analysıs of the electrostatic, dispersion and total energy frameworks augmented stabilization through electrostatic energy contribution. Moreover, optimized molecular structure, using DFT at B3LYP/6–311G(d,p) level was compared with the experimentally determined one. HOMO–LUMO energy gaps were determined and the molecular electrostatic potential (MESP) surfaces are calculated at B3LYP/6–31 G level to predict possible sites for electrophilic and nucleophilic attacks. DFT computations, through stable terminal HOMO and whole molecule LUMO spread, and significant polarity predicted amphoteric behavior of the synthesized 1-(4-methylbenzoyl)thiourea for its reaction with an electrophile or nucleophile.
期刊介绍:
Chemical Data Collections (CDC) provides a publication outlet for the increasing need to make research material and data easy to share and re-use. Publication of research data with CDC will allow scientists to: -Make their data easy to find and access -Benefit from the fast publication process -Contribute to proper data citation and attribution -Publish their intermediate and null/negative results -Receive recognition for the work that does not fit traditional article format. The research data will be published as ''data articles'' that support fast and easy submission and quick peer-review processes. Data articles introduced by CDC are short self-contained publications about research materials and data. They must provide the scientific context of the described work and contain the following elements: a title, list of authors (plus affiliations), abstract, keywords, graphical abstract, metadata table, main text and at least three references. The journal welcomes submissions focusing on (but not limited to) the following categories of research output: spectral data, syntheses, crystallographic data, computational simulations, molecular dynamics and models, physicochemical data, etc.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


