保留在内质网中的内胚层蛋白突变体通过对野生型等位基因产生显性负效应而加剧遗传性出血性毛细血管扩张症1型(HHT1)的功能丧失
IF 2.5
3区 生物学
Q3 CELL BIOLOGY
引用次数: 0
摘要
遗传性出血性毛细血管扩张症(HHT)是一种常染色体显性遗传疾病,每 5000-8000 人中就有 1 人患病。遗传性出血性毛细血管扩张症 1 型(HHT1)是最常见的遗传性出血性毛细血管扩张症,表现为各种血管畸形,从鼻衄、粘膜和皮肤毛细血管扩张症等轻微症状到肺部、脑部或肝部严重的动静脉畸形(AVM)不等。HHT1是由ENG基因的杂合突变引起的,ENG基因编码TGFβ同源二聚体共受体endoglin。以前的研究表明,一些内胚叶蛋白 HHT1 致病变体由于滞留在内质网(ER)中,因此无法转运到质膜,从而被ER相关降解(ERAD)降解。Endoglin 是一种在 ER 中形成的同源二聚体,因此我们假设 ER 保留的变体与 WT 蛋白之间可能会形成混合的异源二聚体,从而阻碍其成熟和向质膜的迁移,造成显性的负面影响。事实上,HA 标记的 ER 保留突变体与 Myc 标记的 WT endoglin 形成了异二聚体。此外,变体 L32R、V105D、P165L、I271N 和 C363Y 通过降低 WT endoglin 的成熟度和质膜定位,对其贩运产生了不利影响。这些结果强烈表明,这些保留ER的变体会产生显性负效应,加剧与WT等位基因杂合表达这些变体的患者的内切蛋白功能丧失。此外,这项研究可能有助于解释在 HHT1 患者中观察到的一些变异性,这些变异除了单倍体功能缺失造成的功能丧失外,显性负效应还造成了额外的功能丧失。这些发现还可能对受ERAD影响的许多疾病产生影响。本文章由计算机程序翻译,如有差异,请以英文原文为准。
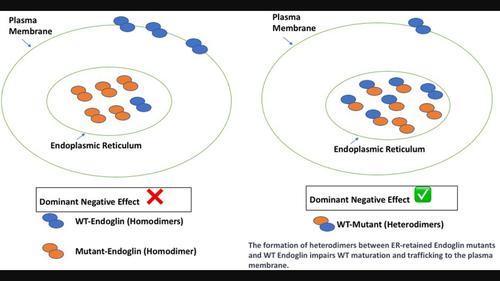
Endoglin mutants retained in the endoplasmic reticulum exacerbate loss of function in hereditary hemorrhagic telangiectasia type 1 (HHT1) by exerting dominant negative effects on the wild type allele
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant disorder affecting 1 in 5000–8000 individuals. Hereditary hemorrhagic telangiectasia type 1 (HHT1) is the most common HHT and manifests as diverse vascular malformations ranging from mild symptoms such as epistaxis and mucosal and cutaneous telangiectases to severe arteriovenous malformations (AVMs) in the lungs, brain or liver. HHT1 is caused by heterozygous mutations in the ENG gene, which encodes endoglin, the TGFβ homodimeric co-receptor. It was previously shown that some endoglin HHT1-causing variants failed to traffic to the plasma membrane due to their retention in the endoplasmic reticulum (ER) and consequent degradation by ER-associated degradation (ERAD). Endoglin is a homodimer formed in the ER, and we therefore hypothesized that mixed heterodimers might form between ER-retained variants and WT protein, thus hampering its maturation and trafficking to the plasma membrane causing dominant negative effects. Indeed, HA-tagged ER-retained mutants formed heterodimers with Myc-tagged WT endoglin. Moreover, variants L32R, V105D, P165L, I271N and C363Y adversely affected the trafficking of WT endoglin by reducing its maturation and plasma membrane localization. These results strongly suggest dominant negative effects exerted by these ER-retained variants aggravating endoglin loss of function in patients expressing them in the heterozygous state with the WT allele. Moreover, this study may help explain some of the variability observed among HHT1 patients due to the additional loss of function exerted by the dominant negative effects in addition to that due to haploinsufficiency. These findings might also have implications for some of the many conditions impacted by ERAD.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Traffic
生物-细胞生物学
CiteScore
8.10
自引率
2.20%
发文量
50
审稿时长
2 months
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


