Zhen Jin, Yadong Han, Jiaxin Zhang, Zhao Liu, Ran Li, Zhao Liu
{"title":"通过与自然杀伤细胞相关的基因特征预测和治疗甲状腺癌","authors":"Zhen Jin, Yadong Han, Jiaxin Zhang, Zhao Liu, Ran Li, Zhao Liu","doi":"10.1002/jgm.3657","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Natural killer (NK) cells are crucial to cancer development and prognosis. However, the role of NK cell-related genes in immunotherapy and the tumor immune microenvironment (TIME) is not well understood. This study aimed to develop reliable risk signatures associated with NK cell-related genes for predicting thyroid cancer (THCA).</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>The single-cell RNA sequencing (scRNA-seq) data from seven THCA samples (GSE184362) and bulk-RNA-seq data of 502 THCA patients (TCGA-THCA) were included. The scRNA-seq data was analyzed using the “Seurat” R package to identify differentially expressed genes in NK cells. The clustering analysis was carried out using the R package “ConsensusClusterPlus”. The gene set variation analysis (GSVA) algorithm was applied to assess the variations in biological pathways among subtypes. The ESTIMATE algorithm was utilized to calculate the scores for stromal, immune and estimate variables. In addition, we used the single sample Gene Set Enrichment Analysis and CIBERSORT algorithms to assess the degree to which immune cells and pathways related to immunity were enriched based on the meta-cohort. In the TCGA-THCA cohort, the “glmnet” R package was used for the gene selection, and LASSO Cox analysis was used to construct prognostic features. The “maftools” R package was used to examine the somatic mutation landscape of THCA in both low- and high-risk groups.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>One-hundred and eighty-five NK cell marker genes were screened, and nine genes were associated with the THCA prognosis. KLF2, OSTF1 and TAPBP were finally identified and constructed a risk signature with significant prognostic value. KLF2 and OSTF1 were protective genes, and TAPBP was a risk gene. Patients at high risk had a considerably lower overall survival compared with those at low risk. Mutations in the TCGA-THCA cohort were predominantly C > T. Increased tumor mutation burden (TMB) levels were linked to overall survival. The low-risk H-TMB+ group had a better prognosis, while the high-risk L-TMB+ group had the worst prognosis.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>Natural killer cell-related genes KLF2, OSTF1 and TAPBP were used to develop a novel prognostic risk signature, offering a new perspective on the prognosis and treatment of THCA.</p>\n </section>\n </div>","PeriodicalId":56122,"journal":{"name":"Journal of Gene Medicine","volume":"26 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-01-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Prognosis and therapy in thyroid cancer by gene signatures related to natural killer cells\",\"authors\":\"Zhen Jin, Yadong Han, Jiaxin Zhang, Zhao Liu, Ran Li, Zhao Liu\",\"doi\":\"10.1002/jgm.3657\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Natural killer (NK) cells are crucial to cancer development and prognosis. However, the role of NK cell-related genes in immunotherapy and the tumor immune microenvironment (TIME) is not well understood. This study aimed to develop reliable risk signatures associated with NK cell-related genes for predicting thyroid cancer (THCA).</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>The single-cell RNA sequencing (scRNA-seq) data from seven THCA samples (GSE184362) and bulk-RNA-seq data of 502 THCA patients (TCGA-THCA) were included. The scRNA-seq data was analyzed using the “Seurat” R package to identify differentially expressed genes in NK cells. The clustering analysis was carried out using the R package “ConsensusClusterPlus”. The gene set variation analysis (GSVA) algorithm was applied to assess the variations in biological pathways among subtypes. The ESTIMATE algorithm was utilized to calculate the scores for stromal, immune and estimate variables. In addition, we used the single sample Gene Set Enrichment Analysis and CIBERSORT algorithms to assess the degree to which immune cells and pathways related to immunity were enriched based on the meta-cohort. In the TCGA-THCA cohort, the “glmnet” R package was used for the gene selection, and LASSO Cox analysis was used to construct prognostic features. The “maftools” R package was used to examine the somatic mutation landscape of THCA in both low- and high-risk groups.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>One-hundred and eighty-five NK cell marker genes were screened, and nine genes were associated with the THCA prognosis. KLF2, OSTF1 and TAPBP were finally identified and constructed a risk signature with significant prognostic value. KLF2 and OSTF1 were protective genes, and TAPBP was a risk gene. Patients at high risk had a considerably lower overall survival compared with those at low risk. Mutations in the TCGA-THCA cohort were predominantly C > T. Increased tumor mutation burden (TMB) levels were linked to overall survival. The low-risk H-TMB+ group had a better prognosis, while the high-risk L-TMB+ group had the worst prognosis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusion</h3>\\n \\n <p>Natural killer cell-related genes KLF2, OSTF1 and TAPBP were used to develop a novel prognostic risk signature, offering a new perspective on the prognosis and treatment of THCA.</p>\\n </section>\\n </div>\",\"PeriodicalId\":56122,\"journal\":{\"name\":\"Journal of Gene Medicine\",\"volume\":\"26 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-01-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Gene Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jgm.3657\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Gene Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jgm.3657","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Prognosis and therapy in thyroid cancer by gene signatures related to natural killer cells
Background
Natural killer (NK) cells are crucial to cancer development and prognosis. However, the role of NK cell-related genes in immunotherapy and the tumor immune microenvironment (TIME) is not well understood. This study aimed to develop reliable risk signatures associated with NK cell-related genes for predicting thyroid cancer (THCA).
Methods
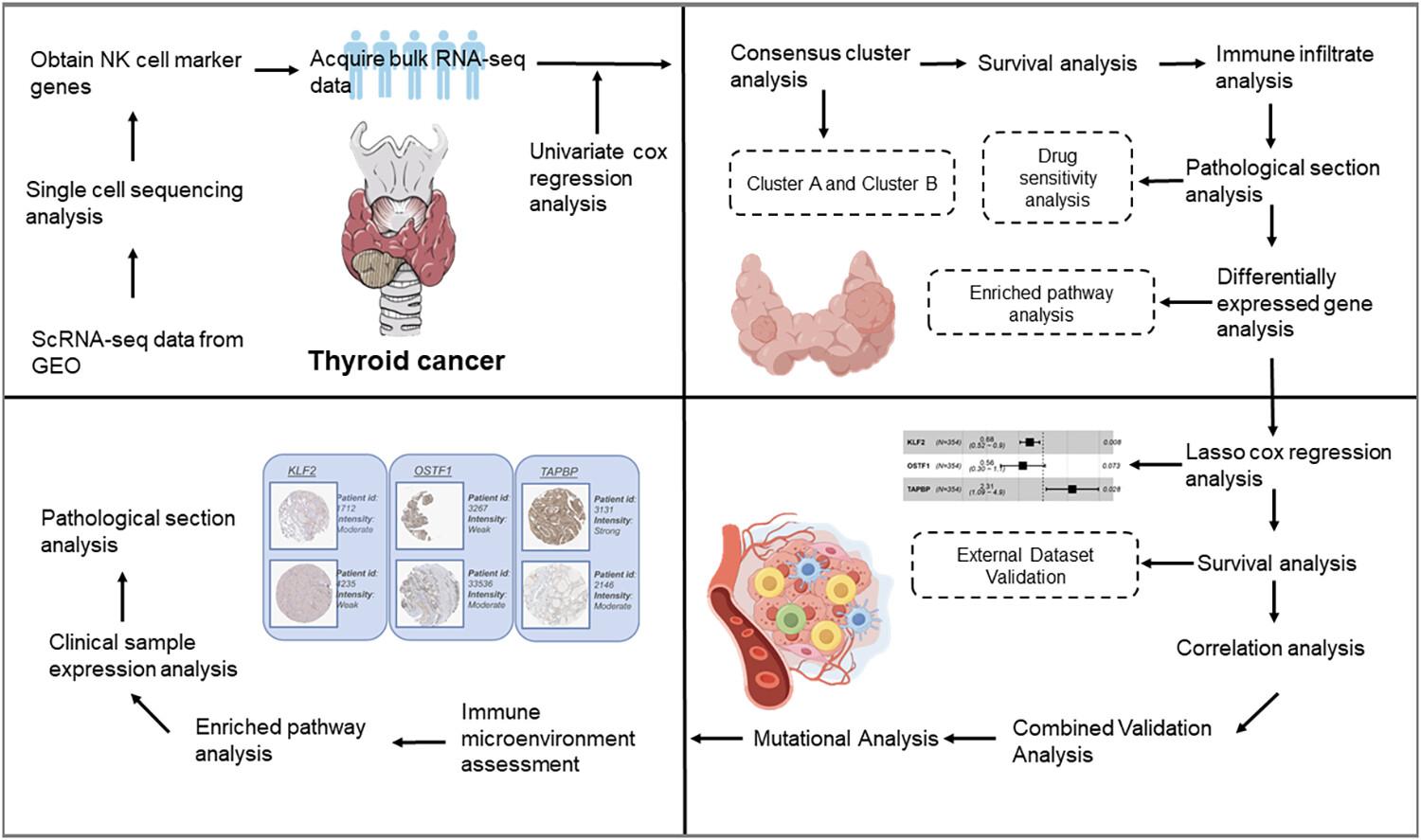
The single-cell RNA sequencing (scRNA-seq) data from seven THCA samples (GSE184362) and bulk-RNA-seq data of 502 THCA patients (TCGA-THCA) were included. The scRNA-seq data was analyzed using the “Seurat” R package to identify differentially expressed genes in NK cells. The clustering analysis was carried out using the R package “ConsensusClusterPlus”. The gene set variation analysis (GSVA) algorithm was applied to assess the variations in biological pathways among subtypes. The ESTIMATE algorithm was utilized to calculate the scores for stromal, immune and estimate variables. In addition, we used the single sample Gene Set Enrichment Analysis and CIBERSORT algorithms to assess the degree to which immune cells and pathways related to immunity were enriched based on the meta-cohort. In the TCGA-THCA cohort, the “glmnet” R package was used for the gene selection, and LASSO Cox analysis was used to construct prognostic features. The “maftools” R package was used to examine the somatic mutation landscape of THCA in both low- and high-risk groups.
Results
One-hundred and eighty-five NK cell marker genes were screened, and nine genes were associated with the THCA prognosis. KLF2, OSTF1 and TAPBP were finally identified and constructed a risk signature with significant prognostic value. KLF2 and OSTF1 were protective genes, and TAPBP was a risk gene. Patients at high risk had a considerably lower overall survival compared with those at low risk. Mutations in the TCGA-THCA cohort were predominantly C > T. Increased tumor mutation burden (TMB) levels were linked to overall survival. The low-risk H-TMB+ group had a better prognosis, while the high-risk L-TMB+ group had the worst prognosis.
Conclusion
Natural killer cell-related genes KLF2, OSTF1 and TAPBP were used to develop a novel prognostic risk signature, offering a new perspective on the prognosis and treatment of THCA.
期刊介绍:
The aims and scope of The Journal of Gene Medicine include cutting-edge science of gene transfer and its applications in gene and cell therapy, genome editing with precision nucleases, epigenetic modifications of host genome by small molecules, siRNA, microRNA and other noncoding RNAs as therapeutic gene-modulating agents or targets, biomarkers for precision medicine, and gene-based prognostic/diagnostic studies.
Key areas of interest are the design of novel synthetic and viral vectors, novel therapeutic nucleic acids such as mRNA, modified microRNAs and siRNAs, antagomirs, aptamers, antisense and exon-skipping agents, refined genome editing tools using nucleic acid /protein combinations, physically or biologically targeted delivery and gene modulation, ex vivo or in vivo pharmacological studies including animal models, and human clinical trials.
Papers presenting research into the mechanisms underlying transfer and action of gene medicines, the application of the new technologies for stem cell modification or nucleic acid based vaccines, the identification of new genetic or epigenetic variations as biomarkers to direct precision medicine, and the preclinical/clinical development of gene/expression signatures indicative of diagnosis or predictive of prognosis are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


