Chinmaya U Joisa, Kevin A Chen, Samantha Beville, Timothy Stuhlmiller, Matthew E Berginski, Denis Okumu, Brian T Golitz, Michael P East, Gary L Johnson, Shawn M Gomez
{"title":"联合激酶组抑制状态可预测癌细胞系对激酶抑制剂联合疗法的敏感性。","authors":"Chinmaya U Joisa, Kevin A Chen, Samantha Beville, Timothy Stuhlmiller, Matthew E Berginski, Denis Okumu, Brian T Golitz, Michael P East, Gary L Johnson, Shawn M Gomez","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p>Protein kinases are a primary focus in targeted therapy development for cancer, owing to their role as regulators in nearly all areas of cell life. Recent strategies targeting the kinome with combination therapies have shown promise, such as trametinib and dabrafenib in advanced melanoma, but empirical design for less characterized pathways remains a challenge. Computational combination screening is an attractive alternative, allowing in-silico filtering prior to experimental testing of drastically fewer leads, increasing efficiency and effectiveness of drug development pipelines. In this work, we generated combined kinome inhibition states of 40,000 kinase inhibitor combinations from kinobeads-based kinome profiling across 64 doses. We then integrated these with transcriptomics from CCLE to build machine learning models with elastic-net feature selection to predict cell line sensitivity across nine cancer types, with accuracy R2 ∼ 0.75-0.9. We then validated the model by using a PDX-derived TNBC cell line and saw good global accuracy (R2 ∼ 0.7) as well as high accuracy in predicting synergy using four popular metrics (R2 ∼ 0.9). Additionally, the model was able to predict a highly synergistic combination of trametinib and omipalisib for TNBC treatment, which incidentally was recently in phase I clinical trials. Our choice of tree-based models for greater interpretability allowed interrogation of highly predictive kinases in each cancer type, such as the MAPK, CDK, and STK kinases. Overall, these results suggest that kinome inhibition states of kinase inhibitor combinations are strongly predictive of cell line responses and have great potential for integration into computational drug screening pipelines. This approach may facilitate the identification of effective kinase inhibitor combinations and accelerate the development of novel cancer therapies, ultimately improving patient outcomes.</p>","PeriodicalId":34954,"journal":{"name":"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing","volume":"29 ","pages":"276-290"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11413988/pdf/","citationCount":"0","resultStr":"{\"title\":\"Combined kinome inhibition states are predictive of cancer cell line sensitivity to kinase inhibitor combination therapies.\",\"authors\":\"Chinmaya U Joisa, Kevin A Chen, Samantha Beville, Timothy Stuhlmiller, Matthew E Berginski, Denis Okumu, Brian T Golitz, Michael P East, Gary L Johnson, Shawn M Gomez\",\"doi\":\"\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Protein kinases are a primary focus in targeted therapy development for cancer, owing to their role as regulators in nearly all areas of cell life. Recent strategies targeting the kinome with combination therapies have shown promise, such as trametinib and dabrafenib in advanced melanoma, but empirical design for less characterized pathways remains a challenge. Computational combination screening is an attractive alternative, allowing in-silico filtering prior to experimental testing of drastically fewer leads, increasing efficiency and effectiveness of drug development pipelines. In this work, we generated combined kinome inhibition states of 40,000 kinase inhibitor combinations from kinobeads-based kinome profiling across 64 doses. We then integrated these with transcriptomics from CCLE to build machine learning models with elastic-net feature selection to predict cell line sensitivity across nine cancer types, with accuracy R2 ∼ 0.75-0.9. We then validated the model by using a PDX-derived TNBC cell line and saw good global accuracy (R2 ∼ 0.7) as well as high accuracy in predicting synergy using four popular metrics (R2 ∼ 0.9). Additionally, the model was able to predict a highly synergistic combination of trametinib and omipalisib for TNBC treatment, which incidentally was recently in phase I clinical trials. Our choice of tree-based models for greater interpretability allowed interrogation of highly predictive kinases in each cancer type, such as the MAPK, CDK, and STK kinases. Overall, these results suggest that kinome inhibition states of kinase inhibitor combinations are strongly predictive of cell line responses and have great potential for integration into computational drug screening pipelines. This approach may facilitate the identification of effective kinase inhibitor combinations and accelerate the development of novel cancer therapies, ultimately improving patient outcomes.</p>\",\"PeriodicalId\":34954,\"journal\":{\"name\":\"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing\",\"volume\":\"29 \",\"pages\":\"276-290\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11413988/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Computer Science\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Computer Science","Score":null,"Total":0}
Combined kinome inhibition states are predictive of cancer cell line sensitivity to kinase inhibitor combination therapies.
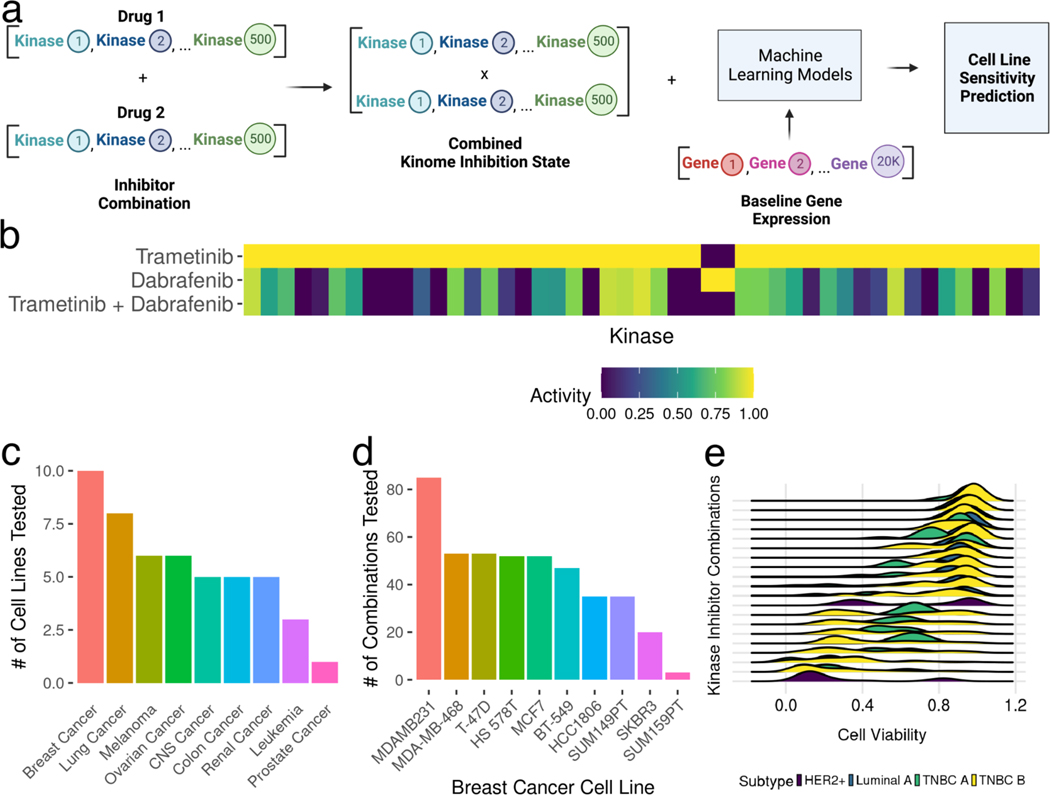
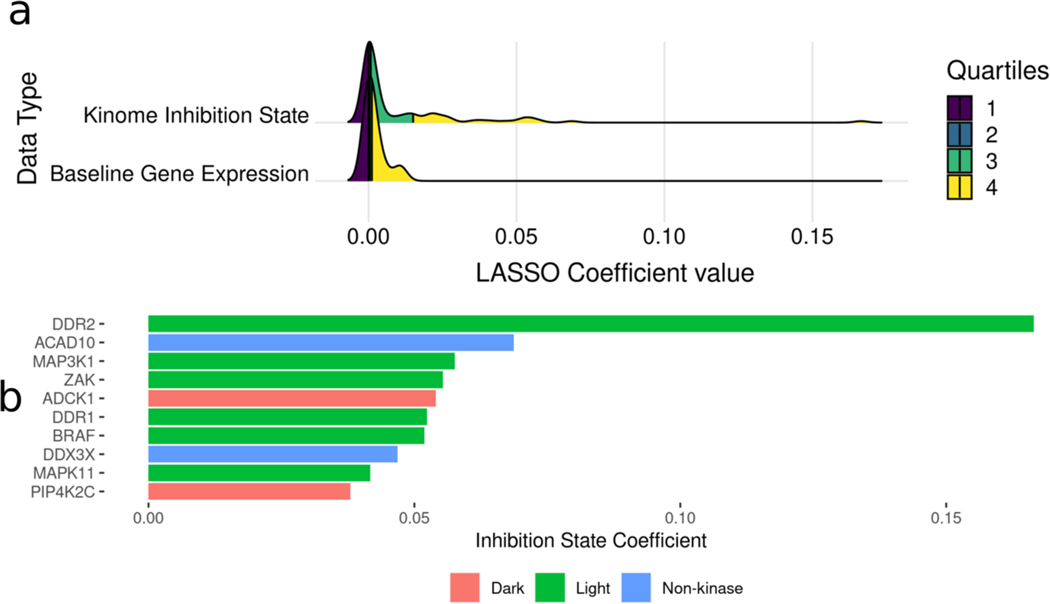
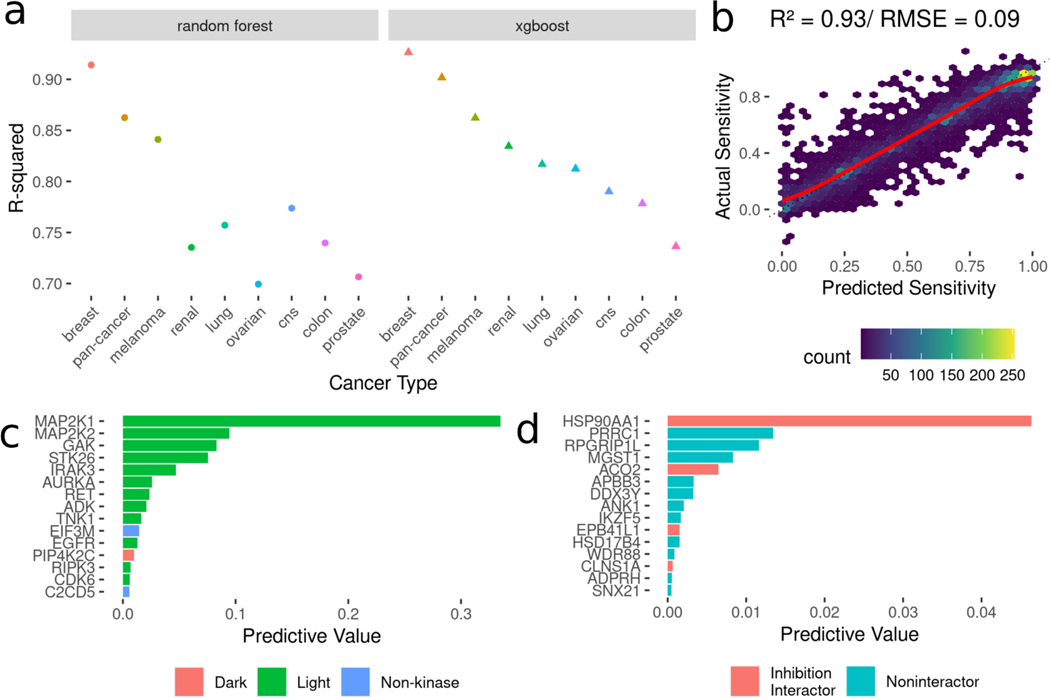
Protein kinases are a primary focus in targeted therapy development for cancer, owing to their role as regulators in nearly all areas of cell life. Recent strategies targeting the kinome with combination therapies have shown promise, such as trametinib and dabrafenib in advanced melanoma, but empirical design for less characterized pathways remains a challenge. Computational combination screening is an attractive alternative, allowing in-silico filtering prior to experimental testing of drastically fewer leads, increasing efficiency and effectiveness of drug development pipelines. In this work, we generated combined kinome inhibition states of 40,000 kinase inhibitor combinations from kinobeads-based kinome profiling across 64 doses. We then integrated these with transcriptomics from CCLE to build machine learning models with elastic-net feature selection to predict cell line sensitivity across nine cancer types, with accuracy R2 ∼ 0.75-0.9. We then validated the model by using a PDX-derived TNBC cell line and saw good global accuracy (R2 ∼ 0.7) as well as high accuracy in predicting synergy using four popular metrics (R2 ∼ 0.9). Additionally, the model was able to predict a highly synergistic combination of trametinib and omipalisib for TNBC treatment, which incidentally was recently in phase I clinical trials. Our choice of tree-based models for greater interpretability allowed interrogation of highly predictive kinases in each cancer type, such as the MAPK, CDK, and STK kinases. Overall, these results suggest that kinome inhibition states of kinase inhibitor combinations are strongly predictive of cell line responses and have great potential for integration into computational drug screening pipelines. This approach may facilitate the identification of effective kinase inhibitor combinations and accelerate the development of novel cancer therapies, ultimately improving patient outcomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


