Somayeh Rahmati, Mojgan Ayoubi-Chianeh, Mohamad Z. Kassaee
{"title":"14 族取代的芽烯对二氢的活化作用","authors":"Somayeh Rahmati, Mojgan Ayoubi-Chianeh, Mohamad Z. Kassaee","doi":"10.1002/poc.4593","DOIUrl":null,"url":null,"abstract":"<p>We have investigated the structural and thermodynamic parameters of group-14 substituted germylenes and their reactivity toward the H<sub>2</sub> molecule using density functional theory (DFT). We conducted the detailed Kohn–Sham molecular orbital (KS-MO) analysis to quantify the effective factors behind the increased reactivity of germylenes in going from C to Sn as substituents. The quantum theory of atoms in molecules (QTAIM), non-covalent interaction (NCI), and natural bond orbital (NBO) analyses revealed the nature of bonds and interactions and demonstrated the reactivity trend of germylenes in the presence of H<sub>2</sub>. The results showed that in going from C to Sn, the reactivity increased due to an improvement in \n<math>\n <mi>σ</mi></math>-donation interaction between the filled lone-pair orbital of the germylene (LP<sub>Ge</sub>) and the \n<math>\n <mi>σ</mi></math>*-orbital of H<sub>2</sub>, which decreased the reaction barrier (\n<math>\n <mo>∆</mo></math><i>E</i><sup>‡</sup>). As the germylene substitution was varied from C to Sn, a significant reactivity was observed for the germylene toward the H<sub>2</sub>. This observation was caused by a reduction in steric repulsion between the germylene and the H<sub>2</sub> and less activation energy due to the higher \n<math>\n <mi>σ</mi></math>-donation and lower back-donation. We have presented the reactivity of new and rationally designed germylenes toward H<sub>2</sub> using various analyses that will serve as a guide for the activation of small molecules such as H<sub>2</sub>, which is employed in many subsequent reactions.</p>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"37 3","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2023-12-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Activation of dihydrogen by group-14 substituted germylenes\",\"authors\":\"Somayeh Rahmati, Mojgan Ayoubi-Chianeh, Mohamad Z. Kassaee\",\"doi\":\"10.1002/poc.4593\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>We have investigated the structural and thermodynamic parameters of group-14 substituted germylenes and their reactivity toward the H<sub>2</sub> molecule using density functional theory (DFT). We conducted the detailed Kohn–Sham molecular orbital (KS-MO) analysis to quantify the effective factors behind the increased reactivity of germylenes in going from C to Sn as substituents. The quantum theory of atoms in molecules (QTAIM), non-covalent interaction (NCI), and natural bond orbital (NBO) analyses revealed the nature of bonds and interactions and demonstrated the reactivity trend of germylenes in the presence of H<sub>2</sub>. The results showed that in going from C to Sn, the reactivity increased due to an improvement in \\n<math>\\n <mi>σ</mi></math>-donation interaction between the filled lone-pair orbital of the germylene (LP<sub>Ge</sub>) and the \\n<math>\\n <mi>σ</mi></math>*-orbital of H<sub>2</sub>, which decreased the reaction barrier (\\n<math>\\n <mo>∆</mo></math><i>E</i><sup>‡</sup>). As the germylene substitution was varied from C to Sn, a significant reactivity was observed for the germylene toward the H<sub>2</sub>. This observation was caused by a reduction in steric repulsion between the germylene and the H<sub>2</sub> and less activation energy due to the higher \\n<math>\\n <mi>σ</mi></math>-donation and lower back-donation. We have presented the reactivity of new and rationally designed germylenes toward H<sub>2</sub> using various analyses that will serve as a guide for the activation of small molecules such as H<sub>2</sub>, which is employed in many subsequent reactions.</p>\",\"PeriodicalId\":16829,\"journal\":{\"name\":\"Journal of Physical Organic Chemistry\",\"volume\":\"37 3\",\"pages\":\"\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2023-12-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physical Organic Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/poc.4593\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4593","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
引用次数: 0
摘要
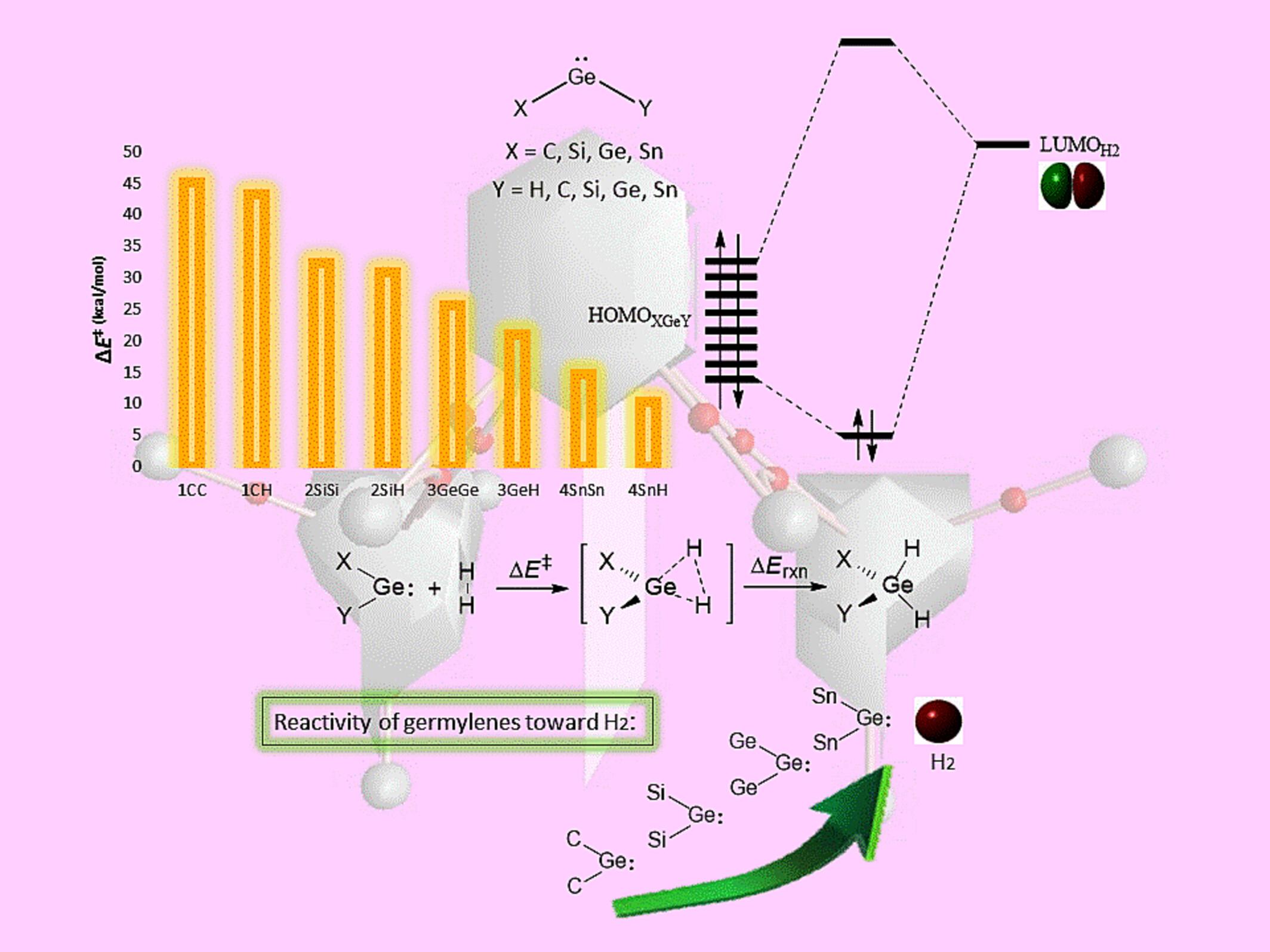
我们利用密度泛函理论(DFT)研究了 14 族取代的萜烯的结构和热力学参数及其对 H2 分子的反应性。我们进行了详细的 Kohn-Sham 分子轨道 (KS-MO) 分析,以量化取代基从 C 变为 Sn 时芽烯反应性提高背后的有效因素。分子中原子的量子理论(QTAIM)、非共价相互作用(NCI)和天然键轨道(NBO)分析揭示了键和相互作用的性质,并证明了在 H2 存在下芽烯的反应性趋势。结果表明,在从 C 到 Sn 的过程中,反应活性增加了,这是由于胚芽烯(LPGe)的填充单对轨道与 H2 的 σ$$ \sigma $$* 轨道之间的 σ$$ \sigma $$--donation 相互作用得到了改善,从而降低了反应障碍(∆$$ \Delta $$E‡)。随着胚芽烯取代物从 C 到 Sn 的变化,观察到胚芽烯对 H2 有显著的反应活性。这一观察结果是由亚胚芽烯与 H2 之间的立体排斥减少以及活化能降低引起的,活化能降低的原因是较高的σ$$ \sigma $$-donation 和较低的 back-donation。我们利用各种分析方法介绍了新的和合理设计的亚芽烯对 H2 的反应性,这些分析方法将为 H2 等小分子的活化提供指导,H2 在许多后续反应中都会用到。
Activation of dihydrogen by group-14 substituted germylenes
We have investigated the structural and thermodynamic parameters of group-14 substituted germylenes and their reactivity toward the H2 molecule using density functional theory (DFT). We conducted the detailed Kohn–Sham molecular orbital (KS-MO) analysis to quantify the effective factors behind the increased reactivity of germylenes in going from C to Sn as substituents. The quantum theory of atoms in molecules (QTAIM), non-covalent interaction (NCI), and natural bond orbital (NBO) analyses revealed the nature of bonds and interactions and demonstrated the reactivity trend of germylenes in the presence of H2. The results showed that in going from C to Sn, the reactivity increased due to an improvement in
-donation interaction between the filled lone-pair orbital of the germylene (LPGe) and the
*-orbital of H2, which decreased the reaction barrier (
E‡). As the germylene substitution was varied from C to Sn, a significant reactivity was observed for the germylene toward the H2. This observation was caused by a reduction in steric repulsion between the germylene and the H2 and less activation energy due to the higher
-donation and lower back-donation. We have presented the reactivity of new and rationally designed germylenes toward H2 using various analyses that will serve as a guide for the activation of small molecules such as H2, which is employed in many subsequent reactions.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


