{"title":"使用氧化石墨烯纳米片从水溶液中去除二苯甲酮污染物的理论见解","authors":"Samaneh Fazli, Foad Buazar, Abdolkarim Matroudi","doi":"10.1007/s00214-023-03076-8","DOIUrl":null,"url":null,"abstract":"<p>In this study, we investigate the adsorption behavior of three benzophenone derivatives, namely 2,4-Dihydroxybenzophenone (benzophenone-1; BP-1), 2,2′,4,4′-tetrahydroxybenzophenone (benzophenone-2; BP-2), and 2-hydroxy-4-methoxybenzophenone (benzophenone-3; BP-3) on the surfaces of graphene oxide (GO) using density functional theory method. The geometric optimization of the unaltered structures of GO adsorbent, benzophenone derivatives, and their respective complexes was conducted via the M052X/6-311 + G* level of theory. The optimal temperature for the interaction between the GO adsorbent and BPs pollutants in the aqueous phase was found to be 298.15 K. The adsorption process was found to be spontaneous, exothermic, and irreversible based on the calculated values of adsorption energy, Gibbs free energy (Δ<i>G</i><sub>ad</sub>), and enthalpy (Δ<i>H</i><sub>ad</sub>). The negative values of the calculated chemical potential for all structures indicated that the studied structures were thermodynamically stable. The adsorption of BP-2 pollutant on the surface of GO results in a highest dipole moment (<i>μ</i><sub><i>d</i></sub> = 28.22 D) compared to the corresponding unadsorbed molecule (<i>μ</i><sub><i>d</i></sub> = 7.93 D). The adsorption efficiency of GO–BPs complexes follows an increasing trend of GO–BP-2 (−1009.75 kcal/mol) > GO–BP-3 (−1006.31 kcal/mol) > GO–BP-1(−1000.65 kcal/mol). Moreover, infrared (IR) frequency calculations confirmed the feasibility of the structures, showing true local minima. The recovery time values indicate that GO is a highly effective adsorbent in removing organic BP-2 pollutants (<span>\\(\\tau =\\)</span> 3.158 ms) from aqueous media rather than BP-3 (<span>\\(\\tau =\\)</span> 2.120 ms) and BP-1 (<span>\\(\\tau =\\)</span> 1.831 ms) counterparts. Other key parameters engaged in the adsorption behavior of considered molecules, including charge capacity, electrophilicity, band gap, chemical potential, and chemical hardness, were also deliberated.</p>","PeriodicalId":23045,"journal":{"name":"Theoretical Chemistry Accounts","volume":"54 10","pages":""},"PeriodicalIF":1.5000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theoretical insights into benzophenone pollutants removal from aqueous solutions using graphene oxide nanosheets\",\"authors\":\"Samaneh Fazli, Foad Buazar, Abdolkarim Matroudi\",\"doi\":\"10.1007/s00214-023-03076-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In this study, we investigate the adsorption behavior of three benzophenone derivatives, namely 2,4-Dihydroxybenzophenone (benzophenone-1; BP-1), 2,2′,4,4′-tetrahydroxybenzophenone (benzophenone-2; BP-2), and 2-hydroxy-4-methoxybenzophenone (benzophenone-3; BP-3) on the surfaces of graphene oxide (GO) using density functional theory method. The geometric optimization of the unaltered structures of GO adsorbent, benzophenone derivatives, and their respective complexes was conducted via the M052X/6-311 + G* level of theory. The optimal temperature for the interaction between the GO adsorbent and BPs pollutants in the aqueous phase was found to be 298.15 K. The adsorption process was found to be spontaneous, exothermic, and irreversible based on the calculated values of adsorption energy, Gibbs free energy (Δ<i>G</i><sub>ad</sub>), and enthalpy (Δ<i>H</i><sub>ad</sub>). The negative values of the calculated chemical potential for all structures indicated that the studied structures were thermodynamically stable. The adsorption of BP-2 pollutant on the surface of GO results in a highest dipole moment (<i>μ</i><sub><i>d</i></sub> = 28.22 D) compared to the corresponding unadsorbed molecule (<i>μ</i><sub><i>d</i></sub> = 7.93 D). The adsorption efficiency of GO–BPs complexes follows an increasing trend of GO–BP-2 (−1009.75 kcal/mol) > GO–BP-3 (−1006.31 kcal/mol) > GO–BP-1(−1000.65 kcal/mol). Moreover, infrared (IR) frequency calculations confirmed the feasibility of the structures, showing true local minima. The recovery time values indicate that GO is a highly effective adsorbent in removing organic BP-2 pollutants (<span>\\\\(\\\\tau =\\\\)</span> 3.158 ms) from aqueous media rather than BP-3 (<span>\\\\(\\\\tau =\\\\)</span> 2.120 ms) and BP-1 (<span>\\\\(\\\\tau =\\\\)</span> 1.831 ms) counterparts. Other key parameters engaged in the adsorption behavior of considered molecules, including charge capacity, electrophilicity, band gap, chemical potential, and chemical hardness, were also deliberated.</p>\",\"PeriodicalId\":23045,\"journal\":{\"name\":\"Theoretical Chemistry Accounts\",\"volume\":\"54 10\",\"pages\":\"\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Theoretical Chemistry Accounts\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1007/s00214-023-03076-8\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Chemistry Accounts","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1007/s00214-023-03076-8","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Theoretical insights into benzophenone pollutants removal from aqueous solutions using graphene oxide nanosheets
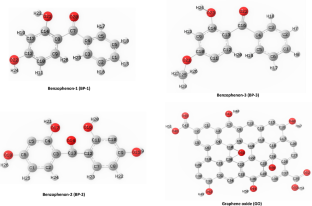
In this study, we investigate the adsorption behavior of three benzophenone derivatives, namely 2,4-Dihydroxybenzophenone (benzophenone-1; BP-1), 2,2′,4,4′-tetrahydroxybenzophenone (benzophenone-2; BP-2), and 2-hydroxy-4-methoxybenzophenone (benzophenone-3; BP-3) on the surfaces of graphene oxide (GO) using density functional theory method. The geometric optimization of the unaltered structures of GO adsorbent, benzophenone derivatives, and their respective complexes was conducted via the M052X/6-311 + G* level of theory. The optimal temperature for the interaction between the GO adsorbent and BPs pollutants in the aqueous phase was found to be 298.15 K. The adsorption process was found to be spontaneous, exothermic, and irreversible based on the calculated values of adsorption energy, Gibbs free energy (ΔGad), and enthalpy (ΔHad). The negative values of the calculated chemical potential for all structures indicated that the studied structures were thermodynamically stable. The adsorption of BP-2 pollutant on the surface of GO results in a highest dipole moment (μd = 28.22 D) compared to the corresponding unadsorbed molecule (μd = 7.93 D). The adsorption efficiency of GO–BPs complexes follows an increasing trend of GO–BP-2 (−1009.75 kcal/mol) > GO–BP-3 (−1006.31 kcal/mol) > GO–BP-1(−1000.65 kcal/mol). Moreover, infrared (IR) frequency calculations confirmed the feasibility of the structures, showing true local minima. The recovery time values indicate that GO is a highly effective adsorbent in removing organic BP-2 pollutants (\(\tau =\) 3.158 ms) from aqueous media rather than BP-3 (\(\tau =\) 2.120 ms) and BP-1 (\(\tau =\) 1.831 ms) counterparts. Other key parameters engaged in the adsorption behavior of considered molecules, including charge capacity, electrophilicity, band gap, chemical potential, and chemical hardness, were also deliberated.
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


