Anna Abramova, Mahdi Rivandi, Liwen Yang, Nadia Stamm, Jan-Philipp Cieslik, Ellen Honisch, Dieter Niederacher, Tanja Fehm, Hans Neubauer, André Franken
{"title":"用于 RNA 测序分析的非凋亡单个循环肿瘤细胞的富集、鉴定和分离工作流程。","authors":"Anna Abramova, Mahdi Rivandi, Liwen Yang, Nadia Stamm, Jan-Philipp Cieslik, Ellen Honisch, Dieter Niederacher, Tanja Fehm, Hans Neubauer, André Franken","doi":"10.1002/cyto.a.24816","DOIUrl":null,"url":null,"abstract":"<p>Circulating tumor cells (CTCs) are constantly shed by tumor tissue and can serve as a valuable analyte for a gene expression analysis from a liquid biopsy. However, a high proportion of CTCs can be apoptotic leading to rapid mRNA decay and challenging the analysis of their transcriptome. We established a workflow to enrich, to identify, and to isolate single CTCs including the discrimination of apoptotic and non-apoptotic CTCs for further single CTC transcriptome analysis. Viable tumor cells—we first used cells from breast cancer cell lines followed by CTCs from metastatic breast cancer patients—were enriched with the CellSearch system from diagnostic leukapheresis products, identified by immunofluorescence analysis for neoplastic markers, and isolated by micromanipulation. Then, their cDNA was generated, amplified, and sequenced. In order to exclude early apoptotic tumor cells, staining with Annexin V coupled to a fluorescent dye was used. Annexin V staining intensity was associated with decreased RNA integrity as well as lower numbers of total reads, exon reads, and detected genes in cell line cells and CTCs. A comparative RNA analysis of single cells from MDA-MB-231 and MCF7 cell lines revealed the expected differential transcriptome profiles. Enrichment and staining procedures of cell line cells that were spiked into blood had only little effect on the obtained RNA sequencing data compared to processing of naïve cells. Further, the detection of transcripts of housekeeping genes such as GAPDH was associated with a significantly higher quality of expression data from CTCs. This workflow enables the enrichment, detection, and isolation of single CTCs for individual transcriptome analyses. The discrimination of apoptotic and non-apoptotic cells allows to focus on CTCs with a high RNA integrity to ensure a successful transcriptome analysis.</p>","PeriodicalId":11068,"journal":{"name":"Cytometry Part A","volume":"105 4","pages":"242-251"},"PeriodicalIF":2.5000,"publicationDate":"2023-12-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cyto.a.24816","citationCount":"0","resultStr":"{\"title\":\"A workflow for the enrichment, the identification, and the isolation of non-apoptotic single circulating tumor cells for RNA sequencing analysis\",\"authors\":\"Anna Abramova, Mahdi Rivandi, Liwen Yang, Nadia Stamm, Jan-Philipp Cieslik, Ellen Honisch, Dieter Niederacher, Tanja Fehm, Hans Neubauer, André Franken\",\"doi\":\"10.1002/cyto.a.24816\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Circulating tumor cells (CTCs) are constantly shed by tumor tissue and can serve as a valuable analyte for a gene expression analysis from a liquid biopsy. However, a high proportion of CTCs can be apoptotic leading to rapid mRNA decay and challenging the analysis of their transcriptome. We established a workflow to enrich, to identify, and to isolate single CTCs including the discrimination of apoptotic and non-apoptotic CTCs for further single CTC transcriptome analysis. Viable tumor cells—we first used cells from breast cancer cell lines followed by CTCs from metastatic breast cancer patients—were enriched with the CellSearch system from diagnostic leukapheresis products, identified by immunofluorescence analysis for neoplastic markers, and isolated by micromanipulation. Then, their cDNA was generated, amplified, and sequenced. In order to exclude early apoptotic tumor cells, staining with Annexin V coupled to a fluorescent dye was used. Annexin V staining intensity was associated with decreased RNA integrity as well as lower numbers of total reads, exon reads, and detected genes in cell line cells and CTCs. A comparative RNA analysis of single cells from MDA-MB-231 and MCF7 cell lines revealed the expected differential transcriptome profiles. Enrichment and staining procedures of cell line cells that were spiked into blood had only little effect on the obtained RNA sequencing data compared to processing of naïve cells. Further, the detection of transcripts of housekeeping genes such as GAPDH was associated with a significantly higher quality of expression data from CTCs. This workflow enables the enrichment, detection, and isolation of single CTCs for individual transcriptome analyses. The discrimination of apoptotic and non-apoptotic cells allows to focus on CTCs with a high RNA integrity to ensure a successful transcriptome analysis.</p>\",\"PeriodicalId\":11068,\"journal\":{\"name\":\"Cytometry Part A\",\"volume\":\"105 4\",\"pages\":\"242-251\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-12-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cyto.a.24816\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cytometry Part A\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/cyto.a.24816\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cytometry Part A","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cyto.a.24816","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
A workflow for the enrichment, the identification, and the isolation of non-apoptotic single circulating tumor cells for RNA sequencing analysis
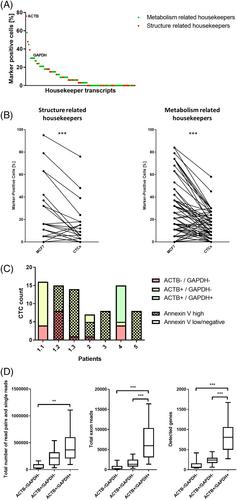
Circulating tumor cells (CTCs) are constantly shed by tumor tissue and can serve as a valuable analyte for a gene expression analysis from a liquid biopsy. However, a high proportion of CTCs can be apoptotic leading to rapid mRNA decay and challenging the analysis of their transcriptome. We established a workflow to enrich, to identify, and to isolate single CTCs including the discrimination of apoptotic and non-apoptotic CTCs for further single CTC transcriptome analysis. Viable tumor cells—we first used cells from breast cancer cell lines followed by CTCs from metastatic breast cancer patients—were enriched with the CellSearch system from diagnostic leukapheresis products, identified by immunofluorescence analysis for neoplastic markers, and isolated by micromanipulation. Then, their cDNA was generated, amplified, and sequenced. In order to exclude early apoptotic tumor cells, staining with Annexin V coupled to a fluorescent dye was used. Annexin V staining intensity was associated with decreased RNA integrity as well as lower numbers of total reads, exon reads, and detected genes in cell line cells and CTCs. A comparative RNA analysis of single cells from MDA-MB-231 and MCF7 cell lines revealed the expected differential transcriptome profiles. Enrichment and staining procedures of cell line cells that were spiked into blood had only little effect on the obtained RNA sequencing data compared to processing of naïve cells. Further, the detection of transcripts of housekeeping genes such as GAPDH was associated with a significantly higher quality of expression data from CTCs. This workflow enables the enrichment, detection, and isolation of single CTCs for individual transcriptome analyses. The discrimination of apoptotic and non-apoptotic cells allows to focus on CTCs with a high RNA integrity to ensure a successful transcriptome analysis.
期刊介绍:
Cytometry Part A, the journal of quantitative single-cell analysis, features original research reports and reviews of innovative scientific studies employing quantitative single-cell measurement, separation, manipulation, and modeling techniques, as well as original articles on mechanisms of molecular and cellular functions obtained by cytometry techniques.
The journal welcomes submissions from multiple research fields that fully embrace the study of the cytome:
Biomedical Instrumentation Engineering
Biophotonics
Bioinformatics
Cell Biology
Computational Biology
Data Science
Immunology
Parasitology
Microbiology
Neuroscience
Cancer
Stem Cells
Tissue Regeneration.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


