Daniel E. Schäffer, Wenrui Li, Abdurrahman Elbasir, Dario C. Altieri, Qi Long, Noam Auslander
{"title":"健康和癌性食道的微生物基因表达分析揭示了临床结果的细菌生物标志物。","authors":"Daniel E. Schäffer, Wenrui Li, Abdurrahman Elbasir, Dario C. Altieri, Qi Long, Noam Auslander","doi":"10.1038/s43705-023-00338-1","DOIUrl":null,"url":null,"abstract":"Local microbiome shifts are implicated in the development and progression of gastrointestinal cancers, and in particular, esophageal carcinoma (ESCA), which is among the most aggressive malignancies. Short-read RNA sequencing (RNAseq) is currently the leading technology to study gene expression changes in cancer. However, using RNAseq to study microbial gene expression is challenging. Here, we establish a new tool to efficiently detect viral and bacterial expression in human tissues through RNAseq. This approach employs a neural network to predict reads of likely microbial origin, which are targeted for assembly into longer contigs, improving identification of microbial species and genes. This approach is applied to perform a systematic comparison of bacterial expression in ESCA and healthy esophagi. We uncover bacterial genera that are over or underabundant in ESCA vs healthy esophagi both before and after correction for possible covariates, including patient metadata. However, we find that bacterial taxonomies are not significantly associated with clinical outcomes. Strikingly, in contrast, dozens of microbial proteins were significantly associated with poor patient outcomes and in particular, proteins that perform mitochondrial functions and iron-sulfur coordination. We further demonstrate associations between these microbial proteins and dysregulated host pathways in ESCA patients. Overall, these results suggest possible influences of bacteria on the development of ESCA and uncover new prognostic biomarkers based on microbial genes. In addition, this study provides a framework for the analysis of other human malignancies whose development may be driven by pathogens.","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":" ","pages":"1-11"},"PeriodicalIF":5.1000,"publicationDate":"2023-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10696091/pdf/","citationCount":"0","resultStr":"{\"title\":\"Microbial gene expression analysis of healthy and cancerous esophagus uncovers bacterial biomarkers of clinical outcomes\",\"authors\":\"Daniel E. Schäffer, Wenrui Li, Abdurrahman Elbasir, Dario C. Altieri, Qi Long, Noam Auslander\",\"doi\":\"10.1038/s43705-023-00338-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Local microbiome shifts are implicated in the development and progression of gastrointestinal cancers, and in particular, esophageal carcinoma (ESCA), which is among the most aggressive malignancies. Short-read RNA sequencing (RNAseq) is currently the leading technology to study gene expression changes in cancer. However, using RNAseq to study microbial gene expression is challenging. Here, we establish a new tool to efficiently detect viral and bacterial expression in human tissues through RNAseq. This approach employs a neural network to predict reads of likely microbial origin, which are targeted for assembly into longer contigs, improving identification of microbial species and genes. This approach is applied to perform a systematic comparison of bacterial expression in ESCA and healthy esophagi. We uncover bacterial genera that are over or underabundant in ESCA vs healthy esophagi both before and after correction for possible covariates, including patient metadata. However, we find that bacterial taxonomies are not significantly associated with clinical outcomes. Strikingly, in contrast, dozens of microbial proteins were significantly associated with poor patient outcomes and in particular, proteins that perform mitochondrial functions and iron-sulfur coordination. We further demonstrate associations between these microbial proteins and dysregulated host pathways in ESCA patients. Overall, these results suggest possible influences of bacteria on the development of ESCA and uncover new prognostic biomarkers based on microbial genes. In addition, this study provides a framework for the analysis of other human malignancies whose development may be driven by pathogens.\",\"PeriodicalId\":73516,\"journal\":{\"name\":\"ISME communications\",\"volume\":\" \",\"pages\":\"1-11\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2023-12-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10696091/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISME communications\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43705-023-00338-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ECOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43705-023-00338-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
Microbial gene expression analysis of healthy and cancerous esophagus uncovers bacterial biomarkers of clinical outcomes
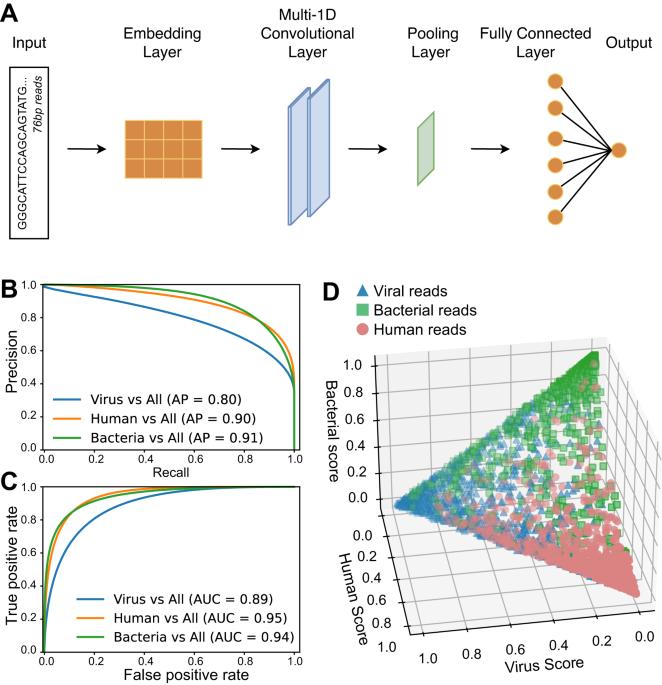
Local microbiome shifts are implicated in the development and progression of gastrointestinal cancers, and in particular, esophageal carcinoma (ESCA), which is among the most aggressive malignancies. Short-read RNA sequencing (RNAseq) is currently the leading technology to study gene expression changes in cancer. However, using RNAseq to study microbial gene expression is challenging. Here, we establish a new tool to efficiently detect viral and bacterial expression in human tissues through RNAseq. This approach employs a neural network to predict reads of likely microbial origin, which are targeted for assembly into longer contigs, improving identification of microbial species and genes. This approach is applied to perform a systematic comparison of bacterial expression in ESCA and healthy esophagi. We uncover bacterial genera that are over or underabundant in ESCA vs healthy esophagi both before and after correction for possible covariates, including patient metadata. However, we find that bacterial taxonomies are not significantly associated with clinical outcomes. Strikingly, in contrast, dozens of microbial proteins were significantly associated with poor patient outcomes and in particular, proteins that perform mitochondrial functions and iron-sulfur coordination. We further demonstrate associations between these microbial proteins and dysregulated host pathways in ESCA patients. Overall, these results suggest possible influences of bacteria on the development of ESCA and uncover new prognostic biomarkers based on microbial genes. In addition, this study provides a framework for the analysis of other human malignancies whose development may be driven by pathogens.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


