{"title":"通过整合大量rna测序和甲基化修饰数据对子宫内膜癌DNA甲基化位点新蓝图的预后评估。","authors":"Huanzhen Zhou, Yingzhi Zhang, Jing Jin, Kewei Shen, Yang Yang, Peiwei Lao","doi":"10.1002/jgm.3638","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Introduction</h3>\n \n <p>Endometrial cancer (EC) is a prevalent malignancy affecting the female population, with an increasing incidence among younger age groups. DNA methylation, a common epigenetic modification, is well-established to play a key role in cancer progression. We suspected whether DNA methylation could be used as biomarkers for EC prognosis.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>In the present study, we analyzed bulk RNA-sequencing data from 544 EC patients and DNA methylation data from 430 EC patients in the TCGA-UCEC cohort. We applied weighted correlation network analysis to select a key gene set associated with panoptosis. We conducted correlation analysis between transcriptomic data of the selected key genes and DNA methylation data to identify valuable DNA methylation sites. These sites were further screened by Cox regression and least absolute shrinkage and selection operator analysis. Immune microenvironment differences between high-risk and low-risk groups were assessed using single-sample gene set enrichment analysi, xCell and MCPcounter algorithms.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Our results identified five DNA methylation sites (cg03906681, cg04549977, cg06029846, cg10043253 and cg15658376) with significant prognostic value in EC. We constructed a prognostic model using these sites, demonstrating satisfactory predictive performance. The low-risk group showed higher immune cell infiltration. Notably, methylation of site cg03906681 was negatively related to CD8 T cell infiltration, whereas cg04549977 exhibited positive correlations with immune infiltration, particularly in macrophages, activated B cells, dendritic cells and myeloid-derived suppressor cells. PD0325901_1060 was strongly correlated with risk scores, indicating a potential therapeutic response for high-risk EC patients.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>We have developed a robust DNA methylation-based prognostic model for EC, which holds promise for improving prognosis prediction and personalized treatment approaches. These findings may contribute to better management of EC patients, particularly in identifying those at higher risk who may benefit from tailored interventions.</p>\n </section>\n </div>","PeriodicalId":56122,"journal":{"name":"Journal of Gene Medicine","volume":null,"pages":null},"PeriodicalIF":3.2000,"publicationDate":"2023-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Prognostic evaluation of the novel blueprint of DNA methylation sites by integrating bulk RNA-sequencing and methylation modification data in endometrial cancer\",\"authors\":\"Huanzhen Zhou, Yingzhi Zhang, Jing Jin, Kewei Shen, Yang Yang, Peiwei Lao\",\"doi\":\"10.1002/jgm.3638\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Introduction</h3>\\n \\n <p>Endometrial cancer (EC) is a prevalent malignancy affecting the female population, with an increasing incidence among younger age groups. DNA methylation, a common epigenetic modification, is well-established to play a key role in cancer progression. We suspected whether DNA methylation could be used as biomarkers for EC prognosis.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>In the present study, we analyzed bulk RNA-sequencing data from 544 EC patients and DNA methylation data from 430 EC patients in the TCGA-UCEC cohort. We applied weighted correlation network analysis to select a key gene set associated with panoptosis. We conducted correlation analysis between transcriptomic data of the selected key genes and DNA methylation data to identify valuable DNA methylation sites. These sites were further screened by Cox regression and least absolute shrinkage and selection operator analysis. Immune microenvironment differences between high-risk and low-risk groups were assessed using single-sample gene set enrichment analysi, xCell and MCPcounter algorithms.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Our results identified five DNA methylation sites (cg03906681, cg04549977, cg06029846, cg10043253 and cg15658376) with significant prognostic value in EC. We constructed a prognostic model using these sites, demonstrating satisfactory predictive performance. The low-risk group showed higher immune cell infiltration. Notably, methylation of site cg03906681 was negatively related to CD8 T cell infiltration, whereas cg04549977 exhibited positive correlations with immune infiltration, particularly in macrophages, activated B cells, dendritic cells and myeloid-derived suppressor cells. PD0325901_1060 was strongly correlated with risk scores, indicating a potential therapeutic response for high-risk EC patients.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusion</h3>\\n \\n <p>We have developed a robust DNA methylation-based prognostic model for EC, which holds promise for improving prognosis prediction and personalized treatment approaches. These findings may contribute to better management of EC patients, particularly in identifying those at higher risk who may benefit from tailored interventions.</p>\\n </section>\\n </div>\",\"PeriodicalId\":56122,\"journal\":{\"name\":\"Journal of Gene Medicine\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-11-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Gene Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jgm.3638\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Gene Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jgm.3638","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
Prognostic evaluation of the novel blueprint of DNA methylation sites by integrating bulk RNA-sequencing and methylation modification data in endometrial cancer
Introduction
Endometrial cancer (EC) is a prevalent malignancy affecting the female population, with an increasing incidence among younger age groups. DNA methylation, a common epigenetic modification, is well-established to play a key role in cancer progression. We suspected whether DNA methylation could be used as biomarkers for EC prognosis.
Methods
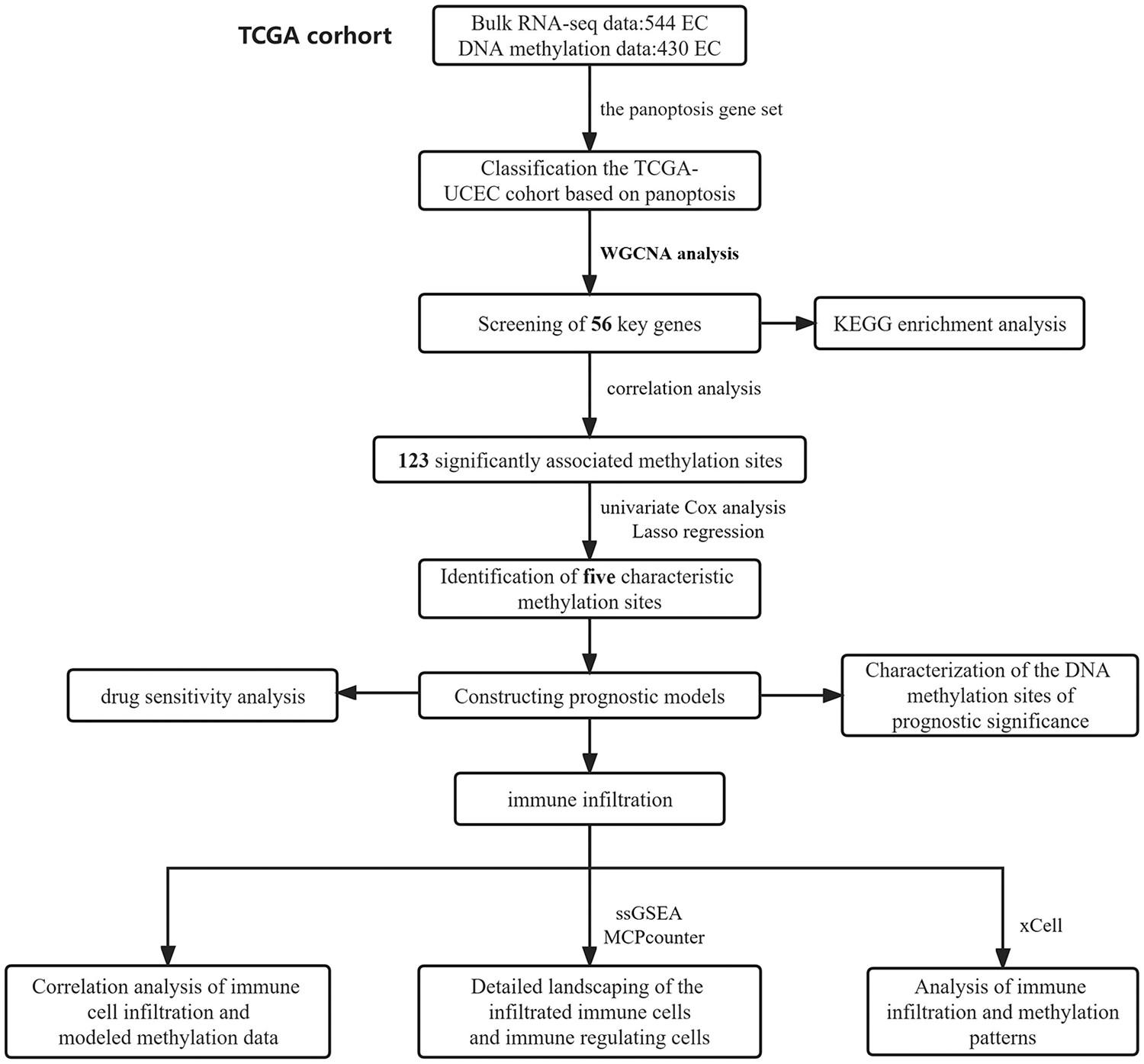
In the present study, we analyzed bulk RNA-sequencing data from 544 EC patients and DNA methylation data from 430 EC patients in the TCGA-UCEC cohort. We applied weighted correlation network analysis to select a key gene set associated with panoptosis. We conducted correlation analysis between transcriptomic data of the selected key genes and DNA methylation data to identify valuable DNA methylation sites. These sites were further screened by Cox regression and least absolute shrinkage and selection operator analysis. Immune microenvironment differences between high-risk and low-risk groups were assessed using single-sample gene set enrichment analysi, xCell and MCPcounter algorithms.
Results
Our results identified five DNA methylation sites (cg03906681, cg04549977, cg06029846, cg10043253 and cg15658376) with significant prognostic value in EC. We constructed a prognostic model using these sites, demonstrating satisfactory predictive performance. The low-risk group showed higher immune cell infiltration. Notably, methylation of site cg03906681 was negatively related to CD8 T cell infiltration, whereas cg04549977 exhibited positive correlations with immune infiltration, particularly in macrophages, activated B cells, dendritic cells and myeloid-derived suppressor cells. PD0325901_1060 was strongly correlated with risk scores, indicating a potential therapeutic response for high-risk EC patients.
Conclusion
We have developed a robust DNA methylation-based prognostic model for EC, which holds promise for improving prognosis prediction and personalized treatment approaches. These findings may contribute to better management of EC patients, particularly in identifying those at higher risk who may benefit from tailored interventions.
期刊介绍:
The aims and scope of The Journal of Gene Medicine include cutting-edge science of gene transfer and its applications in gene and cell therapy, genome editing with precision nucleases, epigenetic modifications of host genome by small molecules, siRNA, microRNA and other noncoding RNAs as therapeutic gene-modulating agents or targets, biomarkers for precision medicine, and gene-based prognostic/diagnostic studies.
Key areas of interest are the design of novel synthetic and viral vectors, novel therapeutic nucleic acids such as mRNA, modified microRNAs and siRNAs, antagomirs, aptamers, antisense and exon-skipping agents, refined genome editing tools using nucleic acid /protein combinations, physically or biologically targeted delivery and gene modulation, ex vivo or in vivo pharmacological studies including animal models, and human clinical trials.
Papers presenting research into the mechanisms underlying transfer and action of gene medicines, the application of the new technologies for stem cell modification or nucleic acid based vaccines, the identification of new genetic or epigenetic variations as biomarkers to direct precision medicine, and the preclinical/clinical development of gene/expression signatures indicative of diagnosis or predictive of prognosis are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


