Irina Zalivina , Temo Barwari , Xiaoke Yin , Sarah R. Langley , Javier Barallobre-Barreiro , Hiroko Wakimoto , Anna Zampetaki , Manuel Mayr , Metin Avkiran , Seda Eminaga
{"title":"在小鼠肥厚性心肌病(HCM)模型中抑制miR-199a-3p可减轻纤维化重塑","authors":"Irina Zalivina , Temo Barwari , Xiaoke Yin , Sarah R. Langley , Javier Barallobre-Barreiro , Hiroko Wakimoto , Anna Zampetaki , Manuel Mayr , Metin Avkiran , Seda Eminaga","doi":"10.1016/j.jmccpl.2023.100056","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disorder, characterized by cardiomyocyte hypertrophy, cardiomyocyte disarray and fibrosis, which has a prevalence of ∼1: 200–500 and predisposes individuals to heart failure and sudden death. The mechanisms through which diverse HCM-causing mutations cause cardiac dysfunction remain mostly unknown and their identification may reveal new therapeutic avenues. MicroRNAs (miRNAs) have emerged as critical regulators of gene expression and disease phenotype in various pathologies. We explored whether miRNAs could play a role in HCM pathogenesis and offer potential therapeutic targets.</p></div><div><h3>Methods and results</h3><p>Using high-throughput miRNA expression profiling and qPCR analysis in two distinct mouse models of HCM, we found that miR-199a-3p expression levels are upregulated in mutant mice compared to age- and treatment-matched wild-type mice. We also found that miR-199a-3p expression is enriched in cardiac non-myocytes compared to cardiomyocytes. When we expressed miR-199a-3p mimic in cultured murine primary cardiac fibroblasts and analyzed the conditioned media by proteomics, we found that several extracellular matrix (ECM) proteins (<em>e.g.</em>, TSP2, FBLN3, COL11A1, LYOX) were differentially secreted (data are available <em>via</em> ProteomeXchange with identifier <span>PXD042904</span><svg><path></path></svg>). We confirmed our proteomics findings by qPCR analysis of selected mRNAs and demonstrated that miR-199a-3p mimic expression in cardiac fibroblasts drives upregulation of ECM gene expression, including <em>Tsp2</em>, <em>Fbln3</em>, <em>Pcoc1</em>, <em>Col1a1</em> and <em>Col3a1</em>. To examine the role of miR-199a-3p <em>in vivo</em>, we inhibited its function using lock-nucleic acid (LNA)-based inhibitors (antimiR-199a-3p) in an HCM mouse model. Our results revealed that progression of cardiac fibrosis is attenuated when miR-199a-3p function is inhibited in mild-to-moderate HCM. Finally, guided by computational target prediction algorithms, we identified mRNAs <em>Cd151</em> and <em>Itga3</em> as direct targets of miR-199a-3p and have shown that miR-199a-3p mimic expression negatively regulates AKT activation in cardiac fibroblasts.</p></div><div><h3>Conclusions</h3><p>Altogether, our results suggest that miR-199a-3p may contribute to cardiac fibrosis in HCM through its actions in cardiac fibroblasts. Thus, inhibition of miR-199a-3p in mild-to-moderate HCM may offer therapeutic benefit in combination with complementary approaches that target the primary defect in cardiac myocytes.</p></div>","PeriodicalId":73835,"journal":{"name":"Journal of molecular and cellular cardiology plus","volume":"6 ","pages":"Article 100056"},"PeriodicalIF":0.0000,"publicationDate":"2023-11-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2772976123000260/pdfft?md5=d7ef72bd8c44be43fadde9936ac62981&pid=1-s2.0-S2772976123000260-main.pdf","citationCount":"1","resultStr":"{\"title\":\"Inhibition of miR-199a-3p in a murine hypertrophic cardiomyopathy (HCM) model attenuates fibrotic remodeling\",\"authors\":\"Irina Zalivina , Temo Barwari , Xiaoke Yin , Sarah R. Langley , Javier Barallobre-Barreiro , Hiroko Wakimoto , Anna Zampetaki , Manuel Mayr , Metin Avkiran , Seda Eminaga\",\"doi\":\"10.1016/j.jmccpl.2023.100056\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disorder, characterized by cardiomyocyte hypertrophy, cardiomyocyte disarray and fibrosis, which has a prevalence of ∼1: 200–500 and predisposes individuals to heart failure and sudden death. The mechanisms through which diverse HCM-causing mutations cause cardiac dysfunction remain mostly unknown and their identification may reveal new therapeutic avenues. MicroRNAs (miRNAs) have emerged as critical regulators of gene expression and disease phenotype in various pathologies. We explored whether miRNAs could play a role in HCM pathogenesis and offer potential therapeutic targets.</p></div><div><h3>Methods and results</h3><p>Using high-throughput miRNA expression profiling and qPCR analysis in two distinct mouse models of HCM, we found that miR-199a-3p expression levels are upregulated in mutant mice compared to age- and treatment-matched wild-type mice. We also found that miR-199a-3p expression is enriched in cardiac non-myocytes compared to cardiomyocytes. When we expressed miR-199a-3p mimic in cultured murine primary cardiac fibroblasts and analyzed the conditioned media by proteomics, we found that several extracellular matrix (ECM) proteins (<em>e.g.</em>, TSP2, FBLN3, COL11A1, LYOX) were differentially secreted (data are available <em>via</em> ProteomeXchange with identifier <span>PXD042904</span><svg><path></path></svg>). We confirmed our proteomics findings by qPCR analysis of selected mRNAs and demonstrated that miR-199a-3p mimic expression in cardiac fibroblasts drives upregulation of ECM gene expression, including <em>Tsp2</em>, <em>Fbln3</em>, <em>Pcoc1</em>, <em>Col1a1</em> and <em>Col3a1</em>. To examine the role of miR-199a-3p <em>in vivo</em>, we inhibited its function using lock-nucleic acid (LNA)-based inhibitors (antimiR-199a-3p) in an HCM mouse model. Our results revealed that progression of cardiac fibrosis is attenuated when miR-199a-3p function is inhibited in mild-to-moderate HCM. Finally, guided by computational target prediction algorithms, we identified mRNAs <em>Cd151</em> and <em>Itga3</em> as direct targets of miR-199a-3p and have shown that miR-199a-3p mimic expression negatively regulates AKT activation in cardiac fibroblasts.</p></div><div><h3>Conclusions</h3><p>Altogether, our results suggest that miR-199a-3p may contribute to cardiac fibrosis in HCM through its actions in cardiac fibroblasts. Thus, inhibition of miR-199a-3p in mild-to-moderate HCM may offer therapeutic benefit in combination with complementary approaches that target the primary defect in cardiac myocytes.</p></div>\",\"PeriodicalId\":73835,\"journal\":{\"name\":\"Journal of molecular and cellular cardiology plus\",\"volume\":\"6 \",\"pages\":\"Article 100056\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-11-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2772976123000260/pdfft?md5=d7ef72bd8c44be43fadde9936ac62981&pid=1-s2.0-S2772976123000260-main.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular and cellular cardiology plus\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2772976123000260\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology plus","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772976123000260","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Inhibition of miR-199a-3p in a murine hypertrophic cardiomyopathy (HCM) model attenuates fibrotic remodeling
Background
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disorder, characterized by cardiomyocyte hypertrophy, cardiomyocyte disarray and fibrosis, which has a prevalence of ∼1: 200–500 and predisposes individuals to heart failure and sudden death. The mechanisms through which diverse HCM-causing mutations cause cardiac dysfunction remain mostly unknown and their identification may reveal new therapeutic avenues. MicroRNAs (miRNAs) have emerged as critical regulators of gene expression and disease phenotype in various pathologies. We explored whether miRNAs could play a role in HCM pathogenesis and offer potential therapeutic targets.
Methods and results
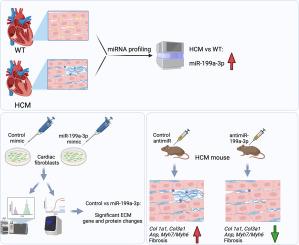
Using high-throughput miRNA expression profiling and qPCR analysis in two distinct mouse models of HCM, we found that miR-199a-3p expression levels are upregulated in mutant mice compared to age- and treatment-matched wild-type mice. We also found that miR-199a-3p expression is enriched in cardiac non-myocytes compared to cardiomyocytes. When we expressed miR-199a-3p mimic in cultured murine primary cardiac fibroblasts and analyzed the conditioned media by proteomics, we found that several extracellular matrix (ECM) proteins (e.g., TSP2, FBLN3, COL11A1, LYOX) were differentially secreted (data are available via ProteomeXchange with identifier PXD042904). We confirmed our proteomics findings by qPCR analysis of selected mRNAs and demonstrated that miR-199a-3p mimic expression in cardiac fibroblasts drives upregulation of ECM gene expression, including Tsp2, Fbln3, Pcoc1, Col1a1 and Col3a1. To examine the role of miR-199a-3p in vivo, we inhibited its function using lock-nucleic acid (LNA)-based inhibitors (antimiR-199a-3p) in an HCM mouse model. Our results revealed that progression of cardiac fibrosis is attenuated when miR-199a-3p function is inhibited in mild-to-moderate HCM. Finally, guided by computational target prediction algorithms, we identified mRNAs Cd151 and Itga3 as direct targets of miR-199a-3p and have shown that miR-199a-3p mimic expression negatively regulates AKT activation in cardiac fibroblasts.
Conclusions
Altogether, our results suggest that miR-199a-3p may contribute to cardiac fibrosis in HCM through its actions in cardiac fibroblasts. Thus, inhibition of miR-199a-3p in mild-to-moderate HCM may offer therapeutic benefit in combination with complementary approaches that target the primary defect in cardiac myocytes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


