{"title":"基于Hellinger距离度量的ATAC-seq数据读取对齐形状景观可视化","authors":"Jian Hao Cheng, Cheng Zheng, Ryo Yamada, Daigo Okada","doi":"10.1111/gtc.13082","DOIUrl":null,"url":null,"abstract":"<p>Assay for Transposase-Accessible Chromatin using high-throughput sequencing (ATAC-seq) is the popular technique using next-generation sequencing to measure chromatin accessibility and identify open chromatin regions. While read alignment shape information of next-generation sequencing data with intensity information has been used in various bioinformatics methods, few studies have focused on pure shape information alone. In this study, we investigated what types of ATAC-seq read alignment shapes are observed for the promoter region and whether the pure shape information was related or unrelated to other gene features. We introduced a novel concept and pipeline for handling the pure shape information of NGS data as probability distributions and quantifying their dissimilarities by information theory. Based on this concept, we demonstrate that the pure shape information of ATAC-seq data is correlated with chromatin openness and some gene characteristics. On the other hand, it is suggested that the pure information of ATAC-seq read alignment shape is unlikely to contain additional information to explain differences in RNA expression. Our study suggests that viewing the read alignment shape of NGS data as probability distributions enables us to capture the characteristics of the genome-wide landscape of such data in a non-parametric manner.</p>","PeriodicalId":12742,"journal":{"name":"Genes to Cells","volume":"29 1","pages":"5-16"},"PeriodicalIF":1.3000,"publicationDate":"2023-11-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Visualization of the landscape of the read alignment shape of ATAC-seq data using Hellinger distance metric\",\"authors\":\"Jian Hao Cheng, Cheng Zheng, Ryo Yamada, Daigo Okada\",\"doi\":\"10.1111/gtc.13082\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Assay for Transposase-Accessible Chromatin using high-throughput sequencing (ATAC-seq) is the popular technique using next-generation sequencing to measure chromatin accessibility and identify open chromatin regions. While read alignment shape information of next-generation sequencing data with intensity information has been used in various bioinformatics methods, few studies have focused on pure shape information alone. In this study, we investigated what types of ATAC-seq read alignment shapes are observed for the promoter region and whether the pure shape information was related or unrelated to other gene features. We introduced a novel concept and pipeline for handling the pure shape information of NGS data as probability distributions and quantifying their dissimilarities by information theory. Based on this concept, we demonstrate that the pure shape information of ATAC-seq data is correlated with chromatin openness and some gene characteristics. On the other hand, it is suggested that the pure information of ATAC-seq read alignment shape is unlikely to contain additional information to explain differences in RNA expression. Our study suggests that viewing the read alignment shape of NGS data as probability distributions enables us to capture the characteristics of the genome-wide landscape of such data in a non-parametric manner.</p>\",\"PeriodicalId\":12742,\"journal\":{\"name\":\"Genes to Cells\",\"volume\":\"29 1\",\"pages\":\"5-16\"},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2023-11-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genes to Cells\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/gtc.13082\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genes to Cells","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/gtc.13082","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Visualization of the landscape of the read alignment shape of ATAC-seq data using Hellinger distance metric
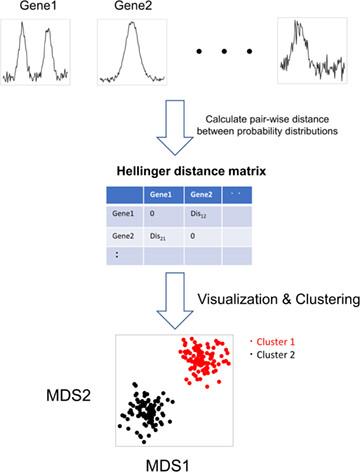
Assay for Transposase-Accessible Chromatin using high-throughput sequencing (ATAC-seq) is the popular technique using next-generation sequencing to measure chromatin accessibility and identify open chromatin regions. While read alignment shape information of next-generation sequencing data with intensity information has been used in various bioinformatics methods, few studies have focused on pure shape information alone. In this study, we investigated what types of ATAC-seq read alignment shapes are observed for the promoter region and whether the pure shape information was related or unrelated to other gene features. We introduced a novel concept and pipeline for handling the pure shape information of NGS data as probability distributions and quantifying their dissimilarities by information theory. Based on this concept, we demonstrate that the pure shape information of ATAC-seq data is correlated with chromatin openness and some gene characteristics. On the other hand, it is suggested that the pure information of ATAC-seq read alignment shape is unlikely to contain additional information to explain differences in RNA expression. Our study suggests that viewing the read alignment shape of NGS data as probability distributions enables us to capture the characteristics of the genome-wide landscape of such data in a non-parametric manner.
期刊介绍:
Genes to Cells provides an international forum for the publication of papers describing important aspects of molecular and cellular biology. The journal aims to present papers that provide conceptual advance in the relevant field. Particular emphasis will be placed on work aimed at understanding the basic mechanisms underlying biological events.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


