丙醛分解机理研究:速率常数评估及动力学模拟
IF 1.5
4区 化学
Q4 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
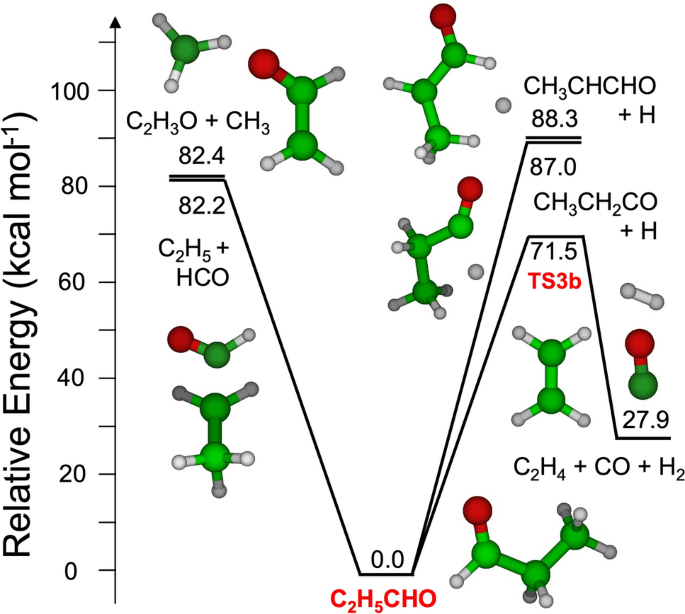
醛类化合物的反应性一直是化学动力学研究的热点,丙醛类化合物通常被认为是其代表性物质。尽管丙醛的反应性具有一定的相关性,但目前对丙醛反应性的估计主要是通过在有限的温度和压力范围内测量的实验数据进行类比和拟合,而少数文献的理论研究更多地集中在势能面(PES)的探索上,而不是对速率常数的估计。本工作的目的是利用从头算过渡态理论为基础的主方程方法重新研究丙烷分解动力学,目的是:(1)确定准确的关键反应通道速率常数;(2)通过模拟现有丙烷热解实验数据,对已有的动力学模型进行了更新和验证。研究发现,丙烷在热解初期通过4个单分子无障碍反应生成CHO + c2h5、ch2 CHO + ch3、ch3 CHCHO + H和ch3 ch2 CO + H,并通过一个三分子途径生成c2h4 + CO + h2。利用变反应坐标过渡态理论确定了每个无障碍反应通道的高压速率常数,并利用整个PES的一维主方程来估计与温度和压力相关的现象通道特定速率常数。这样确定的分解速率常数与少数可用的实验数据一致,并且比以前的文献估计要快得多。最后将估计的动力学参数应用到CRECK动力学机制中,从而与文献中的激波管热解数据更加吻合。本文章由计算机程序翻译,如有差异,请以英文原文为准。
On the decomposition mechanism of propanal: rate constants evaluation and kinetic simulations
Abstract The reactivity of aldehydes has been the subject of considerable interest in chemical kinetics, with propanal often chosen as the representative species. Despite its relevance, the reactivity of propanal is currently estimated from analogy and fitting of experimental data measured in limited temperature and pressure ranges, while the few literature theoretical studies have focused more on the exploration the potential energy surface (PES) than on the estimation of rate constants. The purpose of this work is to reinvestigate the propanal decomposition kinetics using the ab initio transition state theory based master equation approach with the intent of: (1) Determining accurate rate constants of key reaction channels; (2) Updating and validating an existing kinetic model by simulating available experimental data on propanal pyrolysis. It is found that propanal decomposition at the initial stages of pyrolysis occurs through four unimolecular barrierless reactions to form CHO + C 2 H 5 , CH 2 CHO + CH 3 , CH 3 CHCHO + H, and CH 3 CH 2 CO + H, and a termolecular pathway leading to the formation of C 2 H 4 + CO + H 2 . High pressure rate constants were determined for each barrierless reaction channel using Variable Reaction Coordinate Transition State Theory and used to estimate phenomenological temperature and pressure dependent channel specific rate constants integrating the 1 dimensional master equation over the whole PES. The decomposition rate constants so determined are in agreement with the few available experimental data and significantly faster than previous literature estimates. The estimated kinetic parameters were finally implemented into the CRECK kinetic mechanism, leading to an improved agreement with shock tube pyrolysis data from the literature.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Theoretical Chemistry Accounts
化学-物理化学
CiteScore
3.40
自引率
0.00%
发文量
74
审稿时长
3.8 months
期刊介绍:
TCA publishes papers in all fields of theoretical chemistry, computational chemistry, and modeling. Fundamental studies as well as applications are included in the scope. In many cases, theorists and computational chemists have special concerns which reach either across the vertical borders of the special disciplines in chemistry or else across the horizontal borders of structure, spectra, synthesis, and dynamics. TCA is especially interested in papers that impact upon multiple chemical disciplines.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


