{"title":"山茶科山茶基因组调查及SSR分析。","authors":"Yu Bai, Lin Ye, Kang Yang, Hui Wang","doi":"10.1155/2022/5417970","DOIUrl":null,"url":null,"abstract":"<p><p><i>Camellia nitidissima</i> Chi (CNC), a species of golden <i>Camellia</i>, is well known as \"the queen of camellias.\" It is an ornamental, medicinal, and edible plant grown in China. In this study, we conducted a genome survey sequencing analysis and simple sequence repeat (SSR) identification of CNC using the Illumina sequencing platform. The 21-mer analysis predicted its genome size to be 2,778.82 Mb, with heterozygosity and repetition rates of 1.42% and 65.27%, respectively. The CNC genome sequences were assembled into 9,399,197 scaffolds, covering ∼2,910 Mb and an N50 of 869 base pair. Its genomic characteristics were found to be similar to those of <i>Camellia oleifera</i>. In addition, 1,940,616 SSRs were identified from the genome data, including mono-(61.85%), di-(28.71%), tri-(6.51%), tetra-(1.85%), penta-(0.57%), and hexanucleotide motifs (0.51%). We believe these data will provide a useful foundation for the development of novel molecular markers for CNC as well as for further whole-genome sequencing of CNC.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2022 ","pages":"5417970"},"PeriodicalIF":1.4000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9646326/pdf/","citationCount":"1","resultStr":"{\"title\":\"Genome Survey and SSR Analysis of <i>Camellia nitidissima</i> Chi (Theaceae).\",\"authors\":\"Yu Bai, Lin Ye, Kang Yang, Hui Wang\",\"doi\":\"10.1155/2022/5417970\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><i>Camellia nitidissima</i> Chi (CNC), a species of golden <i>Camellia</i>, is well known as \\\"the queen of camellias.\\\" It is an ornamental, medicinal, and edible plant grown in China. In this study, we conducted a genome survey sequencing analysis and simple sequence repeat (SSR) identification of CNC using the Illumina sequencing platform. The 21-mer analysis predicted its genome size to be 2,778.82 Mb, with heterozygosity and repetition rates of 1.42% and 65.27%, respectively. The CNC genome sequences were assembled into 9,399,197 scaffolds, covering ∼2,910 Mb and an N50 of 869 base pair. Its genomic characteristics were found to be similar to those of <i>Camellia oleifera</i>. In addition, 1,940,616 SSRs were identified from the genome data, including mono-(61.85%), di-(28.71%), tri-(6.51%), tetra-(1.85%), penta-(0.57%), and hexanucleotide motifs (0.51%). We believe these data will provide a useful foundation for the development of novel molecular markers for CNC as well as for further whole-genome sequencing of CNC.</p>\",\"PeriodicalId\":12778,\"journal\":{\"name\":\"Genetics research\",\"volume\":\"2022 \",\"pages\":\"5417970\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9646326/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1155/2022/5417970\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2022/5417970","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genome Survey and SSR Analysis of Camellia nitidissima Chi (Theaceae).
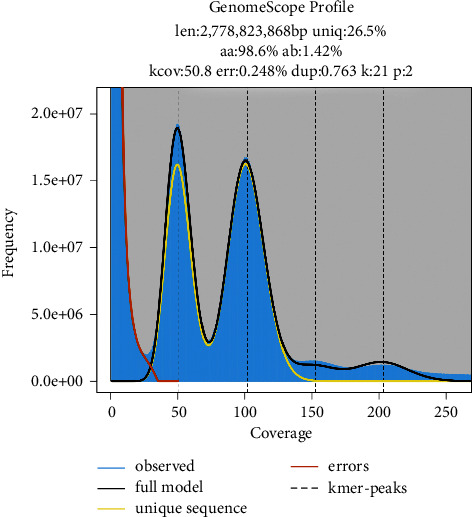
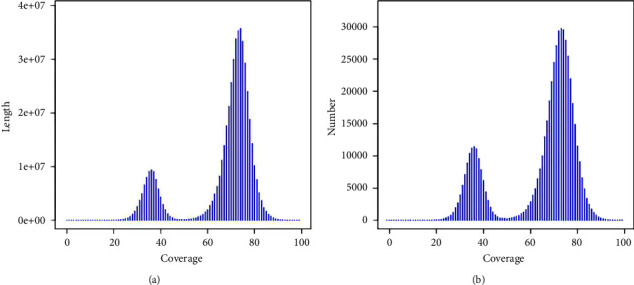
Camellia nitidissima Chi (CNC), a species of golden Camellia, is well known as "the queen of camellias." It is an ornamental, medicinal, and edible plant grown in China. In this study, we conducted a genome survey sequencing analysis and simple sequence repeat (SSR) identification of CNC using the Illumina sequencing platform. The 21-mer analysis predicted its genome size to be 2,778.82 Mb, with heterozygosity and repetition rates of 1.42% and 65.27%, respectively. The CNC genome sequences were assembled into 9,399,197 scaffolds, covering ∼2,910 Mb and an N50 of 869 base pair. Its genomic characteristics were found to be similar to those of Camellia oleifera. In addition, 1,940,616 SSRs were identified from the genome data, including mono-(61.85%), di-(28.71%), tri-(6.51%), tetra-(1.85%), penta-(0.57%), and hexanucleotide motifs (0.51%). We believe these data will provide a useful foundation for the development of novel molecular markers for CNC as well as for further whole-genome sequencing of CNC.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


