Marco Lecis, Katia Rossi, Maria Elena Guerzoni, Ilaria Mariotti, Lorenzo Iughetti
{"title":"酶替代疗法(ERT)对心功能的影响改变了婴儿起病庞贝病患者的预后:家族史","authors":"Marco Lecis, Katia Rossi, Maria Elena Guerzoni, Ilaria Mariotti, Lorenzo Iughetti","doi":"10.1155/2023/8470341","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lysosomal acid alpha-glucosidase (GAA) deficiency, also known as Pompe disease, is an autosomal recessive disorder that leads to the accumulation of glycogen in lysosomes and cytoplasm, resulting in tissue destruction. Infantile-onset GAA deficiency is characterized by cardiomyopathy and severe generalized hypotonia. Without treatment, most patients die within the first two years of life. The demonstration of reduced GAA activity, followed by sequencing of the GAA gene, confirms the disease. GAA deficiency is currently treated with enzyme replacement therapy (ERT) with improved clinical outcomes and survival. <i>Case Presentation</i>. We describe the case of DGAA in two siblings, in which the diagnostic time point, treatment, and outcomes were completely different. The girl was diagnosed with DGAA at the age of 6 months during investigations for poor weight gain and excessive sleepiness. The finding of severe cardiomyopathy through EKG and echocardiography led to the suspicion of storage disease, and the GAA deficiency was later confirmed by genetic analysis. The girl died of complications due to the clinical picture before starting ERT. Conversely, her younger brother had the opportunity to receive an early diagnosis and the rapid onset of ERT. He is showing a regression of cardiac hypertrophy.</p><p><strong>Conclusion: </strong>The advent of ERT improved clinical outcomes and survival in infantile-onset PD. Its impact on cardiac function is still under study, but different reports in the literature have shown encouraging data. Early recognition of DGAA and prompt initiation of ERT is therefore crucial to prevent the progression of the disease and improve the outcomes.</p>","PeriodicalId":9623,"journal":{"name":"Case Reports in Pediatrics","volume":"2023 ","pages":"8470341"},"PeriodicalIF":0.5000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9957634/pdf/","citationCount":"0","resultStr":"{\"title\":\"Enzyme Replacement Therapy (ERT) on Heart Function Changes the Outcome in Patients with Infantile-Onset Pompe Disease: A Familial History.\",\"authors\":\"Marco Lecis, Katia Rossi, Maria Elena Guerzoni, Ilaria Mariotti, Lorenzo Iughetti\",\"doi\":\"10.1155/2023/8470341\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lysosomal acid alpha-glucosidase (GAA) deficiency, also known as Pompe disease, is an autosomal recessive disorder that leads to the accumulation of glycogen in lysosomes and cytoplasm, resulting in tissue destruction. Infantile-onset GAA deficiency is characterized by cardiomyopathy and severe generalized hypotonia. Without treatment, most patients die within the first two years of life. The demonstration of reduced GAA activity, followed by sequencing of the GAA gene, confirms the disease. GAA deficiency is currently treated with enzyme replacement therapy (ERT) with improved clinical outcomes and survival. <i>Case Presentation</i>. We describe the case of DGAA in two siblings, in which the diagnostic time point, treatment, and outcomes were completely different. The girl was diagnosed with DGAA at the age of 6 months during investigations for poor weight gain and excessive sleepiness. The finding of severe cardiomyopathy through EKG and echocardiography led to the suspicion of storage disease, and the GAA deficiency was later confirmed by genetic analysis. The girl died of complications due to the clinical picture before starting ERT. Conversely, her younger brother had the opportunity to receive an early diagnosis and the rapid onset of ERT. He is showing a regression of cardiac hypertrophy.</p><p><strong>Conclusion: </strong>The advent of ERT improved clinical outcomes and survival in infantile-onset PD. Its impact on cardiac function is still under study, but different reports in the literature have shown encouraging data. Early recognition of DGAA and prompt initiation of ERT is therefore crucial to prevent the progression of the disease and improve the outcomes.</p>\",\"PeriodicalId\":9623,\"journal\":{\"name\":\"Case Reports in Pediatrics\",\"volume\":\"2023 \",\"pages\":\"8470341\"},\"PeriodicalIF\":0.5000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9957634/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/8470341\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/8470341","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PEDIATRICS","Score":null,"Total":0}
Enzyme Replacement Therapy (ERT) on Heart Function Changes the Outcome in Patients with Infantile-Onset Pompe Disease: A Familial History.
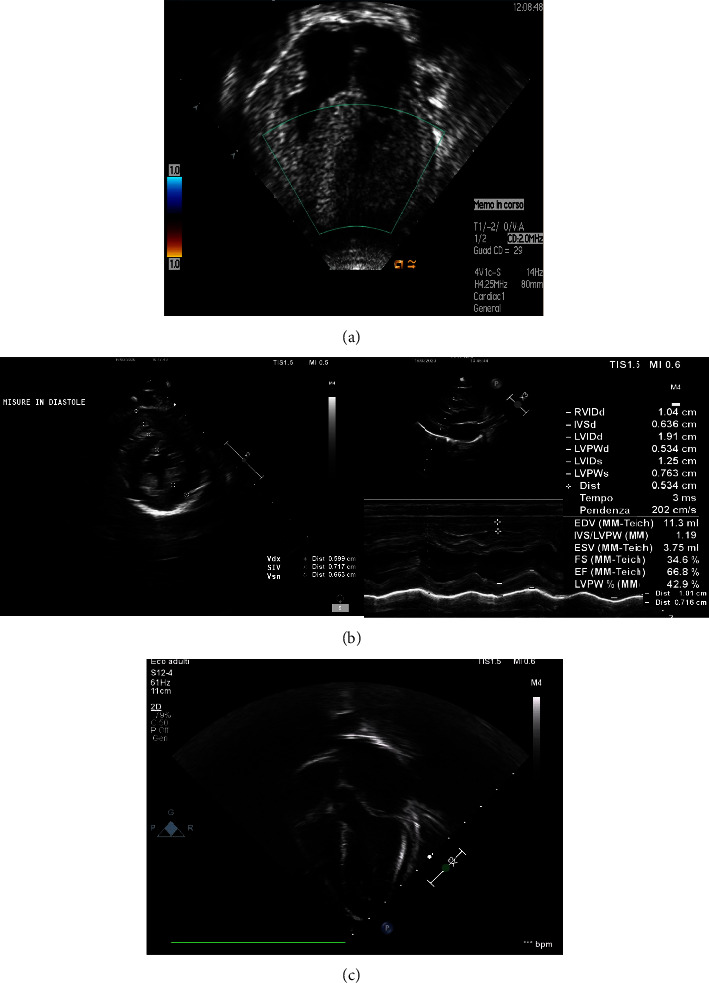
Background: Lysosomal acid alpha-glucosidase (GAA) deficiency, also known as Pompe disease, is an autosomal recessive disorder that leads to the accumulation of glycogen in lysosomes and cytoplasm, resulting in tissue destruction. Infantile-onset GAA deficiency is characterized by cardiomyopathy and severe generalized hypotonia. Without treatment, most patients die within the first two years of life. The demonstration of reduced GAA activity, followed by sequencing of the GAA gene, confirms the disease. GAA deficiency is currently treated with enzyme replacement therapy (ERT) with improved clinical outcomes and survival. Case Presentation. We describe the case of DGAA in two siblings, in which the diagnostic time point, treatment, and outcomes were completely different. The girl was diagnosed with DGAA at the age of 6 months during investigations for poor weight gain and excessive sleepiness. The finding of severe cardiomyopathy through EKG and echocardiography led to the suspicion of storage disease, and the GAA deficiency was later confirmed by genetic analysis. The girl died of complications due to the clinical picture before starting ERT. Conversely, her younger brother had the opportunity to receive an early diagnosis and the rapid onset of ERT. He is showing a regression of cardiac hypertrophy.
Conclusion: The advent of ERT improved clinical outcomes and survival in infantile-onset PD. Its impact on cardiac function is still under study, but different reports in the literature have shown encouraging data. Early recognition of DGAA and prompt initiation of ERT is therefore crucial to prevent the progression of the disease and improve the outcomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


