{"title":"脂肽计算机辅助药物发现的机遇挑战:大分子治疗学的新见解。","authors":"Manisha Yadav, J Satya Eswari","doi":"10.18502/ajmb.v15i1.11419","DOIUrl":null,"url":null,"abstract":"<p><p>Computer-aided drug designing is a promising approach to defeating the dry pipeline of drug discovery. It aims at reduced experimental efforts with cost-effectiveness. Naturally occurring large molecules with molecular weight higher than 500 <i>Dalton</i> such as cationic peptides, cyclic peptides, glycopeptides and lipopeptides are a few examples of large molecules which have successful applications as the broad spectrum antibacterial, anticancer, antiviral, antifungal and antithrombotic drugs. Utilization of microbial metabolites as potential drug candidates incur cost effectiveness through large scale production of such molecules rather than a synthetic approach. Computational studies on such compounds generate tremendous possibilities to develop novel leads with challenges to handle these complex molecules with available computational tools. The opportunities begin with the desired structural modifications in the parent drug molecule. Virtual modifications followed by molecular interaction studies at the target site through molecular modeling simulations and identification of structure-activity relationship models to develop more prominent and potential drug molecules. Lead optimization studies to develop novel compounds with increased specificity and reduced off targeting is a big challenge computationally for large molecules. Prediction of optimized pharmacokinetic properties facilitates development of a compound with lower toxicity as compared to the natural compounds. Generating the library of compounds and studies for target specificity and ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) for large molecules are laborious and incur huge cost and chemical wastage through <i>in-vitro</i> methods. Hence, computational methods need to be explored to develop novel compounds from natural large molecules with higher specificity. This review article is focusing on possible challenges and opportunities in the pathway of computer-aided drug discovery of large molecule therapeutics.</p>","PeriodicalId":8669,"journal":{"name":"Avicenna journal of medical biotechnology","volume":"15 1","pages":"3-13"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ca/42/AJMB-15-3.PMC9895984.pdf","citationCount":"3","resultStr":"{\"title\":\"Opportunistic Challenges of Computer-aided Drug Discovery of Lipopeptides: New Insights for Large Molecule Therapeutics.\",\"authors\":\"Manisha Yadav, J Satya Eswari\",\"doi\":\"10.18502/ajmb.v15i1.11419\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Computer-aided drug designing is a promising approach to defeating the dry pipeline of drug discovery. It aims at reduced experimental efforts with cost-effectiveness. Naturally occurring large molecules with molecular weight higher than 500 <i>Dalton</i> such as cationic peptides, cyclic peptides, glycopeptides and lipopeptides are a few examples of large molecules which have successful applications as the broad spectrum antibacterial, anticancer, antiviral, antifungal and antithrombotic drugs. Utilization of microbial metabolites as potential drug candidates incur cost effectiveness through large scale production of such molecules rather than a synthetic approach. Computational studies on such compounds generate tremendous possibilities to develop novel leads with challenges to handle these complex molecules with available computational tools. The opportunities begin with the desired structural modifications in the parent drug molecule. Virtual modifications followed by molecular interaction studies at the target site through molecular modeling simulations and identification of structure-activity relationship models to develop more prominent and potential drug molecules. Lead optimization studies to develop novel compounds with increased specificity and reduced off targeting is a big challenge computationally for large molecules. Prediction of optimized pharmacokinetic properties facilitates development of a compound with lower toxicity as compared to the natural compounds. Generating the library of compounds and studies for target specificity and ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) for large molecules are laborious and incur huge cost and chemical wastage through <i>in-vitro</i> methods. Hence, computational methods need to be explored to develop novel compounds from natural large molecules with higher specificity. This review article is focusing on possible challenges and opportunities in the pathway of computer-aided drug discovery of large molecule therapeutics.</p>\",\"PeriodicalId\":8669,\"journal\":{\"name\":\"Avicenna journal of medical biotechnology\",\"volume\":\"15 1\",\"pages\":\"3-13\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ca/42/AJMB-15-3.PMC9895984.pdf\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Avicenna journal of medical biotechnology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.18502/ajmb.v15i1.11419\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Avicenna journal of medical biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.18502/ajmb.v15i1.11419","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Opportunistic Challenges of Computer-aided Drug Discovery of Lipopeptides: New Insights for Large Molecule Therapeutics.
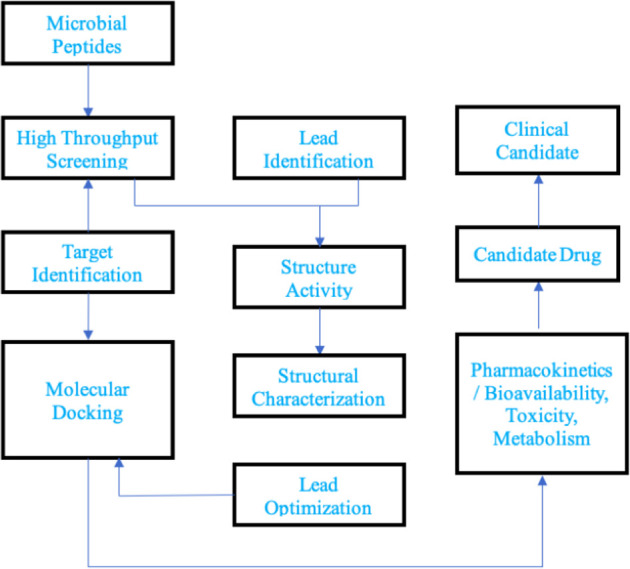
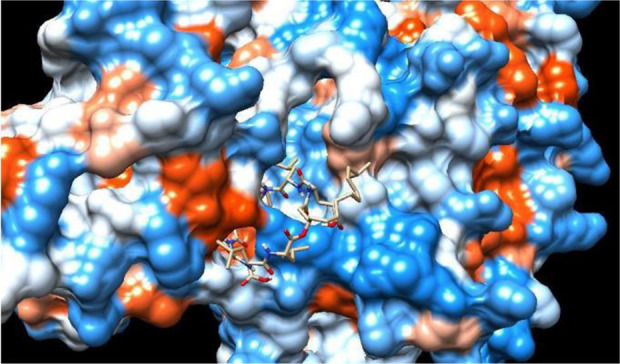
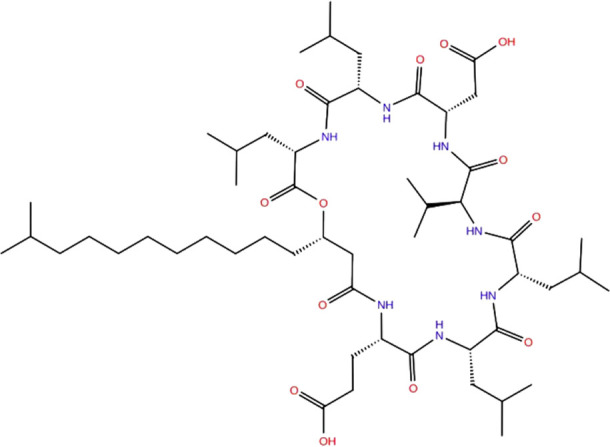
Computer-aided drug designing is a promising approach to defeating the dry pipeline of drug discovery. It aims at reduced experimental efforts with cost-effectiveness. Naturally occurring large molecules with molecular weight higher than 500 Dalton such as cationic peptides, cyclic peptides, glycopeptides and lipopeptides are a few examples of large molecules which have successful applications as the broad spectrum antibacterial, anticancer, antiviral, antifungal and antithrombotic drugs. Utilization of microbial metabolites as potential drug candidates incur cost effectiveness through large scale production of such molecules rather than a synthetic approach. Computational studies on such compounds generate tremendous possibilities to develop novel leads with challenges to handle these complex molecules with available computational tools. The opportunities begin with the desired structural modifications in the parent drug molecule. Virtual modifications followed by molecular interaction studies at the target site through molecular modeling simulations and identification of structure-activity relationship models to develop more prominent and potential drug molecules. Lead optimization studies to develop novel compounds with increased specificity and reduced off targeting is a big challenge computationally for large molecules. Prediction of optimized pharmacokinetic properties facilitates development of a compound with lower toxicity as compared to the natural compounds. Generating the library of compounds and studies for target specificity and ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) for large molecules are laborious and incur huge cost and chemical wastage through in-vitro methods. Hence, computational methods need to be explored to develop novel compounds from natural large molecules with higher specificity. This review article is focusing on possible challenges and opportunities in the pathway of computer-aided drug discovery of large molecule therapeutics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


