Tobias Hoffmann, Nikolaus Gassler, Ulf Teichgräber, Tim Sandhaus, Peter Oelzner, Gunter Wolf, Alexander Pfeil
{"title":"临床影像:Sjögren疾病中的严重间质性肺病-肺部发生了什么?炎症还是纤维化?","authors":"Tobias Hoffmann, Nikolaus Gassler, Ulf Teichgräber, Tim Sandhaus, Peter Oelzner, Gunter Wolf, Alexander Pfeil","doi":"10.1002/acr2.11516","DOIUrl":null,"url":null,"abstract":"The patient, a 66-year-old woman, was fi rst diagnosed with idiopathic pulmonary fi brosis (IPF) in 2015, and therapy with nintedanib was initiated. In 2020, the patient presented to our clinic for differential diagnosis. The laboratory tests revealed an elevated antinuclear antibody titer as well as anti-Ro/SSA and anti-La/SSB antibodies. Lip mucosal biopsy revealed fi ndings of lymphoplasma cellular in fi ltration of the salivary glands consistent with Sjögren disease (SD) ( A-1 and A-2 ). High-resolution computed tomography (HRCT) of the lungs showed marked bronchiectasis, subpleural and basal reticulations, and peripherally basally accentuated ground-glass opacities, but no signi fi cant honeycombing ( B-1 and B-2 ). According to the international IPF guideline, the pattern could be classi fi ed as “ probable usual interstitial pneumonia (UIP) ” (1,2). Because of the patient ’ s poor general health condition, an invasive diagnostic was not possible. Based on these fi ndings, the diagnosis of SD with severe interstitial lung disease (ILD), not responding to nintedanib, was con fi rmed. We then initiated an immunosuppressive induction therapy with cyclophosphamide and glucocorticoids, followed by a switch to mycophenolate mofetil. Due to the rapid and severe pulmonary deterioration, a single lung transplantation was performed. Microscopically, the central and peripheral lung parenchyma revealed a patchy fi brous proliferation with an extensive fi brosis pattern and lymphohistiocytic in fi ltration, especially peripheral with","PeriodicalId":7084,"journal":{"name":"ACR Open Rheumatology","volume":"5 2","pages":"61-62"},"PeriodicalIF":0.0000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/19/2e/ACR2-5-61.PMC9926058.pdf","citationCount":"1","resultStr":"{\"title\":\"Clinical Images: Severe interstitial lung disease in Sjögren disease - What happens in the lungs? Inflammation or fibrosis?\",\"authors\":\"Tobias Hoffmann, Nikolaus Gassler, Ulf Teichgräber, Tim Sandhaus, Peter Oelzner, Gunter Wolf, Alexander Pfeil\",\"doi\":\"10.1002/acr2.11516\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The patient, a 66-year-old woman, was fi rst diagnosed with idiopathic pulmonary fi brosis (IPF) in 2015, and therapy with nintedanib was initiated. In 2020, the patient presented to our clinic for differential diagnosis. The laboratory tests revealed an elevated antinuclear antibody titer as well as anti-Ro/SSA and anti-La/SSB antibodies. Lip mucosal biopsy revealed fi ndings of lymphoplasma cellular in fi ltration of the salivary glands consistent with Sjögren disease (SD) ( A-1 and A-2 ). High-resolution computed tomography (HRCT) of the lungs showed marked bronchiectasis, subpleural and basal reticulations, and peripherally basally accentuated ground-glass opacities, but no signi fi cant honeycombing ( B-1 and B-2 ). According to the international IPF guideline, the pattern could be classi fi ed as “ probable usual interstitial pneumonia (UIP) ” (1,2). Because of the patient ’ s poor general health condition, an invasive diagnostic was not possible. Based on these fi ndings, the diagnosis of SD with severe interstitial lung disease (ILD), not responding to nintedanib, was con fi rmed. We then initiated an immunosuppressive induction therapy with cyclophosphamide and glucocorticoids, followed by a switch to mycophenolate mofetil. Due to the rapid and severe pulmonary deterioration, a single lung transplantation was performed. Microscopically, the central and peripheral lung parenchyma revealed a patchy fi brous proliferation with an extensive fi brosis pattern and lymphohistiocytic in fi ltration, especially peripheral with\",\"PeriodicalId\":7084,\"journal\":{\"name\":\"ACR Open Rheumatology\",\"volume\":\"5 2\",\"pages\":\"61-62\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/19/2e/ACR2-5-61.PMC9926058.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACR Open Rheumatology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1002/acr2.11516\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACR Open Rheumatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/acr2.11516","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
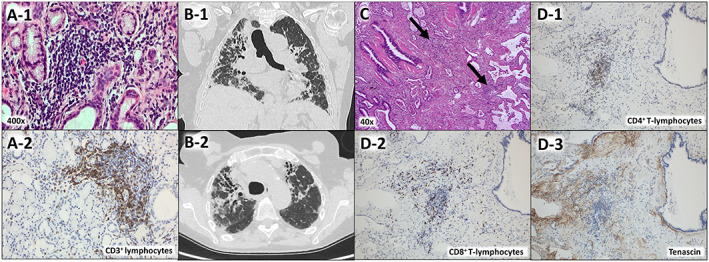
Clinical Images: Severe interstitial lung disease in Sjögren disease - What happens in the lungs? Inflammation or fibrosis?
The patient, a 66-year-old woman, was fi rst diagnosed with idiopathic pulmonary fi brosis (IPF) in 2015, and therapy with nintedanib was initiated. In 2020, the patient presented to our clinic for differential diagnosis. The laboratory tests revealed an elevated antinuclear antibody titer as well as anti-Ro/SSA and anti-La/SSB antibodies. Lip mucosal biopsy revealed fi ndings of lymphoplasma cellular in fi ltration of the salivary glands consistent with Sjögren disease (SD) ( A-1 and A-2 ). High-resolution computed tomography (HRCT) of the lungs showed marked bronchiectasis, subpleural and basal reticulations, and peripherally basally accentuated ground-glass opacities, but no signi fi cant honeycombing ( B-1 and B-2 ). According to the international IPF guideline, the pattern could be classi fi ed as “ probable usual interstitial pneumonia (UIP) ” (1,2). Because of the patient ’ s poor general health condition, an invasive diagnostic was not possible. Based on these fi ndings, the diagnosis of SD with severe interstitial lung disease (ILD), not responding to nintedanib, was con fi rmed. We then initiated an immunosuppressive induction therapy with cyclophosphamide and glucocorticoids, followed by a switch to mycophenolate mofetil. Due to the rapid and severe pulmonary deterioration, a single lung transplantation was performed. Microscopically, the central and peripheral lung parenchyma revealed a patchy fi brous proliferation with an extensive fi brosis pattern and lymphohistiocytic in fi ltration, especially peripheral with

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


