Ashutosh Kumar, Adil Asghar, Himanshu N Singh, Muneeb A Faiq, Sujeet Kumar, Ravi K Narayan, Gopichand Kumar, Prakhar Dwivedi, Chetan Sahni, Rakesh K Jha, Maheswari Kulandhasamy, Pranav Prasoon, Kishore Sesham, Kamla Kant, Sada N Pandey
{"title":"SARS-CoV-2 Omicron 变体基因组序列及其与流行病学的相关性:硅分析。","authors":"Ashutosh Kumar, Adil Asghar, Himanshu N Singh, Muneeb A Faiq, Sujeet Kumar, Ravi K Narayan, Gopichand Kumar, Prakhar Dwivedi, Chetan Sahni, Rakesh K Jha, Maheswari Kulandhasamy, Pranav Prasoon, Kishore Sesham, Kamla Kant, Sada N Pandey","doi":"10.2196/42700","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Emergence of the new SARS-CoV-2 variant B.1.1.529 worried health policy makers worldwide due to a large number of mutations in its genomic sequence, especially in the spike protein region. The World Health Organization (WHO) designated this variant as a global variant of concern (VOC), which was named \"Omicron.\" Following Omicron's emergence, a surge of new COVID-19 cases was reported globally, primarily in South Africa.</p><p><strong>Objective: </strong>The aim of this study was to understand whether Omicron had an epidemiological advantage over existing variants.</p><p><strong>Methods: </strong>We performed an in silico analysis of the complete genomic sequences of Omicron available on the Global Initiative on Sharing Avian Influenza Data (GISAID) database to analyze the functional impact of the mutations present in this variant on virus-host interactions in terms of viral transmissibility, virulence/lethality, and immune escape. In addition, we performed a correlation analysis of the relative proportion of the genomic sequences of specific SARS-CoV-2 variants (in the period from October 1 to November 29, 2021) with matched epidemiological data (new COVID-19 cases and deaths) from South Africa.</p><p><strong>Results: </strong>Compared with the current list of global VOCs/variants of interest (VOIs), as per the WHO, Omicron bears more sequence variation, specifically in the spike protein and host receptor-binding motif (RBM). Omicron showed the closest nucleotide and protein sequence homology with the Alpha variant for the complete sequence and the RBM. The mutations were found to be primarily condensed in the spike region (n=28-48) of the virus. Further mutational analysis showed enrichment for the mutations decreasing binding affinity to angiotensin-converting enzyme 2 receptor and receptor-binding domain protein expression, and for increasing the propensity of immune escape. An inverse correlation of Omicron with the Delta variant was noted (r=-0.99, <i>P</i><.001; 95% CI -0.99 to -0.97) in the sequences reported from South Africa postemergence of the new variant, subsequently showing a decrease. There was a steep rise in new COVID-19 cases in parallel with the increase in the proportion of Omicron isolates since the report of the first case (74%-100%). By contrast, the incidence of new deaths did not increase (r=-0.04, <i>P</i>>.05; 95% CI -0.52 to 0.58).</p><p><strong>Conclusions: </strong>In silico analysis of viral genomic sequences suggests that the Omicron variant has more remarkable immune-escape ability than existing VOCs/VOIs, including Delta, but reduced virulence/lethality than other reported variants. The higher power for immune escape for Omicron was a likely reason for the resurgence in COVID-19 cases and its rapid rise as the globally dominant strain. Being more infectious but less lethal than the existing variants, Omicron could have plausibly led to widespread unnoticed new, repeated, and vaccine breakthrough infections, raising the population-level immunity barrier against the emergence of new lethal variants. The Omicron variant could have thus paved the way for the end of the pandemic.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":"4 ","pages":"e42700"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9843602/pdf/","citationCount":"0","resultStr":"{\"title\":\"SARS-CoV-2 Omicron Variant Genomic Sequences and Their Epidemiological Correlates Regarding the End of the Pandemic: In Silico Analysis.\",\"authors\":\"Ashutosh Kumar, Adil Asghar, Himanshu N Singh, Muneeb A Faiq, Sujeet Kumar, Ravi K Narayan, Gopichand Kumar, Prakhar Dwivedi, Chetan Sahni, Rakesh K Jha, Maheswari Kulandhasamy, Pranav Prasoon, Kishore Sesham, Kamla Kant, Sada N Pandey\",\"doi\":\"10.2196/42700\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Emergence of the new SARS-CoV-2 variant B.1.1.529 worried health policy makers worldwide due to a large number of mutations in its genomic sequence, especially in the spike protein region. The World Health Organization (WHO) designated this variant as a global variant of concern (VOC), which was named \\\"Omicron.\\\" Following Omicron's emergence, a surge of new COVID-19 cases was reported globally, primarily in South Africa.</p><p><strong>Objective: </strong>The aim of this study was to understand whether Omicron had an epidemiological advantage over existing variants.</p><p><strong>Methods: </strong>We performed an in silico analysis of the complete genomic sequences of Omicron available on the Global Initiative on Sharing Avian Influenza Data (GISAID) database to analyze the functional impact of the mutations present in this variant on virus-host interactions in terms of viral transmissibility, virulence/lethality, and immune escape. In addition, we performed a correlation analysis of the relative proportion of the genomic sequences of specific SARS-CoV-2 variants (in the period from October 1 to November 29, 2021) with matched epidemiological data (new COVID-19 cases and deaths) from South Africa.</p><p><strong>Results: </strong>Compared with the current list of global VOCs/variants of interest (VOIs), as per the WHO, Omicron bears more sequence variation, specifically in the spike protein and host receptor-binding motif (RBM). Omicron showed the closest nucleotide and protein sequence homology with the Alpha variant for the complete sequence and the RBM. The mutations were found to be primarily condensed in the spike region (n=28-48) of the virus. Further mutational analysis showed enrichment for the mutations decreasing binding affinity to angiotensin-converting enzyme 2 receptor and receptor-binding domain protein expression, and for increasing the propensity of immune escape. An inverse correlation of Omicron with the Delta variant was noted (r=-0.99, <i>P</i><.001; 95% CI -0.99 to -0.97) in the sequences reported from South Africa postemergence of the new variant, subsequently showing a decrease. There was a steep rise in new COVID-19 cases in parallel with the increase in the proportion of Omicron isolates since the report of the first case (74%-100%). By contrast, the incidence of new deaths did not increase (r=-0.04, <i>P</i>>.05; 95% CI -0.52 to 0.58).</p><p><strong>Conclusions: </strong>In silico analysis of viral genomic sequences suggests that the Omicron variant has more remarkable immune-escape ability than existing VOCs/VOIs, including Delta, but reduced virulence/lethality than other reported variants. The higher power for immune escape for Omicron was a likely reason for the resurgence in COVID-19 cases and its rapid rise as the globally dominant strain. Being more infectious but less lethal than the existing variants, Omicron could have plausibly led to widespread unnoticed new, repeated, and vaccine breakthrough infections, raising the population-level immunity barrier against the emergence of new lethal variants. The Omicron variant could have thus paved the way for the end of the pandemic.</p>\",\"PeriodicalId\":73552,\"journal\":{\"name\":\"JMIR bioinformatics and biotechnology\",\"volume\":\"4 \",\"pages\":\"e42700\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9843602/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JMIR bioinformatics and biotechnology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2196/42700\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/42700","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
SARS-CoV-2 Omicron Variant Genomic Sequences and Their Epidemiological Correlates Regarding the End of the Pandemic: In Silico Analysis.
Background: Emergence of the new SARS-CoV-2 variant B.1.1.529 worried health policy makers worldwide due to a large number of mutations in its genomic sequence, especially in the spike protein region. The World Health Organization (WHO) designated this variant as a global variant of concern (VOC), which was named "Omicron." Following Omicron's emergence, a surge of new COVID-19 cases was reported globally, primarily in South Africa.
Objective: The aim of this study was to understand whether Omicron had an epidemiological advantage over existing variants.
Methods: We performed an in silico analysis of the complete genomic sequences of Omicron available on the Global Initiative on Sharing Avian Influenza Data (GISAID) database to analyze the functional impact of the mutations present in this variant on virus-host interactions in terms of viral transmissibility, virulence/lethality, and immune escape. In addition, we performed a correlation analysis of the relative proportion of the genomic sequences of specific SARS-CoV-2 variants (in the period from October 1 to November 29, 2021) with matched epidemiological data (new COVID-19 cases and deaths) from South Africa.
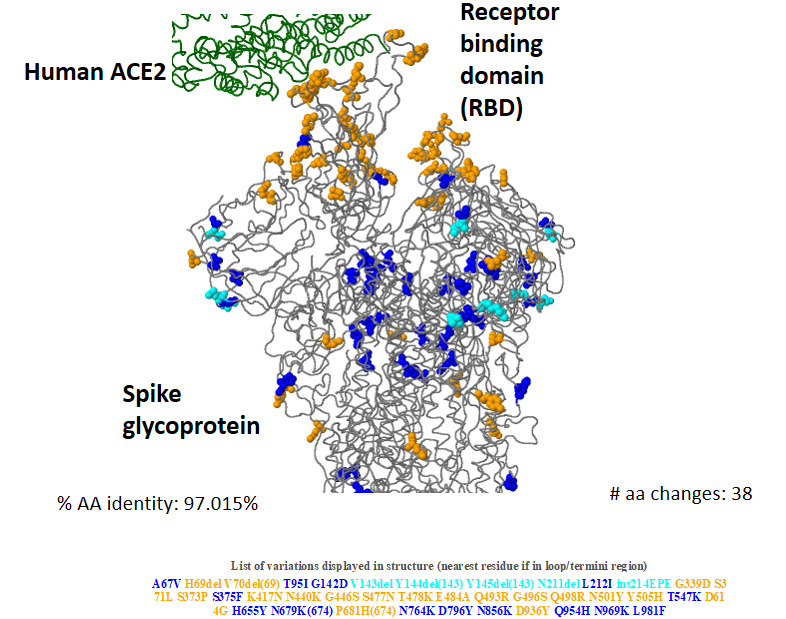
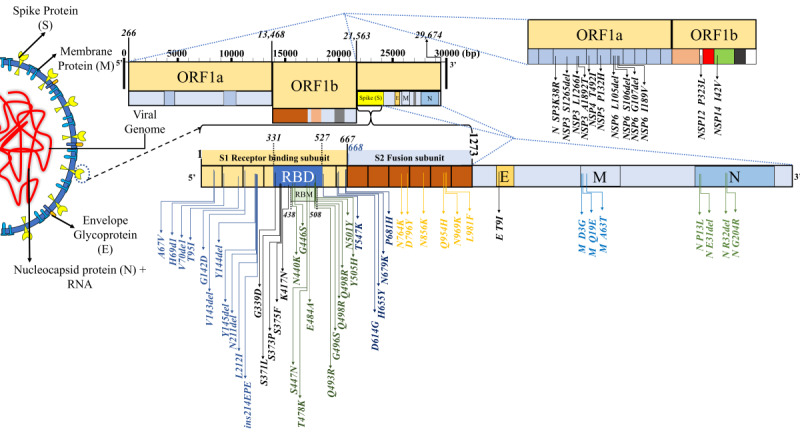
Results: Compared with the current list of global VOCs/variants of interest (VOIs), as per the WHO, Omicron bears more sequence variation, specifically in the spike protein and host receptor-binding motif (RBM). Omicron showed the closest nucleotide and protein sequence homology with the Alpha variant for the complete sequence and the RBM. The mutations were found to be primarily condensed in the spike region (n=28-48) of the virus. Further mutational analysis showed enrichment for the mutations decreasing binding affinity to angiotensin-converting enzyme 2 receptor and receptor-binding domain protein expression, and for increasing the propensity of immune escape. An inverse correlation of Omicron with the Delta variant was noted (r=-0.99, P<.001; 95% CI -0.99 to -0.97) in the sequences reported from South Africa postemergence of the new variant, subsequently showing a decrease. There was a steep rise in new COVID-19 cases in parallel with the increase in the proportion of Omicron isolates since the report of the first case (74%-100%). By contrast, the incidence of new deaths did not increase (r=-0.04, P>.05; 95% CI -0.52 to 0.58).
Conclusions: In silico analysis of viral genomic sequences suggests that the Omicron variant has more remarkable immune-escape ability than existing VOCs/VOIs, including Delta, but reduced virulence/lethality than other reported variants. The higher power for immune escape for Omicron was a likely reason for the resurgence in COVID-19 cases and its rapid rise as the globally dominant strain. Being more infectious but less lethal than the existing variants, Omicron could have plausibly led to widespread unnoticed new, repeated, and vaccine breakthrough infections, raising the population-level immunity barrier against the emergence of new lethal variants. The Omicron variant could have thus paved the way for the end of the pandemic.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


