{"title":"单细胞RNA前mrna测序的长度偏差。","authors":"Gennady Gorin, Lior Pachter","doi":"10.1016/j.bpr.2022.100097","DOIUrl":null,"url":null,"abstract":"<p><p>Single-cell RNA sequencing data can be modeled using Markov chains to yield genome-wide insights into transcriptional physics. However, quantitative inference with such data requires careful assessment of noise sources. We find that long pre-mRNA transcripts are over-represented in sequencing data. To explain this trend, we propose a length-based model of capture bias, which may produce false-positive observations. We solve this model and use it to find concordant parameter trends as well as systematic, mechanistically interpretable technical and biological differences in paired data sets.</p>","PeriodicalId":72402,"journal":{"name":"Biophysical reports","volume":"3 1","pages":"100097"},"PeriodicalIF":2.7000,"publicationDate":"2023-03-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/fb/9b/main.PMC9843228.pdf","citationCount":"16","resultStr":"{\"title\":\"Length biases in single-cell RNA sequencing of pre-mRNA.\",\"authors\":\"Gennady Gorin, Lior Pachter\",\"doi\":\"10.1016/j.bpr.2022.100097\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Single-cell RNA sequencing data can be modeled using Markov chains to yield genome-wide insights into transcriptional physics. However, quantitative inference with such data requires careful assessment of noise sources. We find that long pre-mRNA transcripts are over-represented in sequencing data. To explain this trend, we propose a length-based model of capture bias, which may produce false-positive observations. We solve this model and use it to find concordant parameter trends as well as systematic, mechanistically interpretable technical and biological differences in paired data sets.</p>\",\"PeriodicalId\":72402,\"journal\":{\"name\":\"Biophysical reports\",\"volume\":\"3 1\",\"pages\":\"100097\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2023-03-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/fb/9b/main.PMC9843228.pdf\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biophysical reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1016/j.bpr.2022.100097\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOPHYSICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.bpr.2022.100097","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOPHYSICS","Score":null,"Total":0}
Length biases in single-cell RNA sequencing of pre-mRNA.
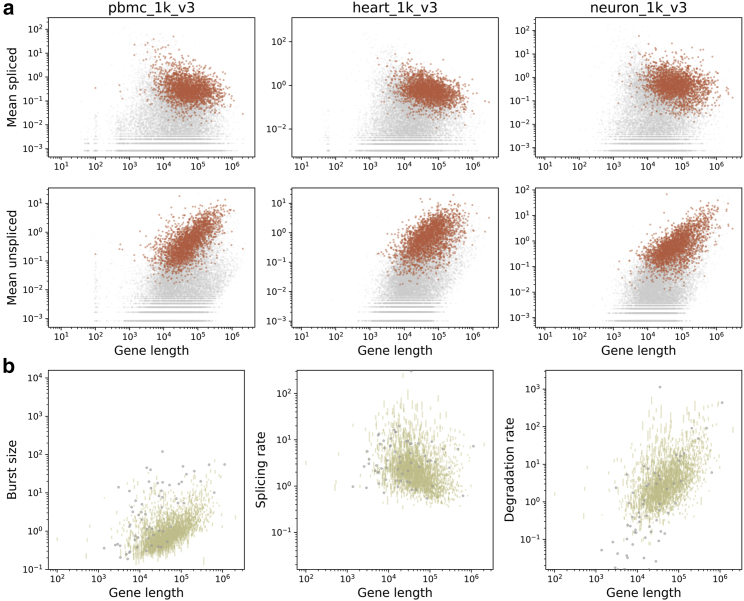
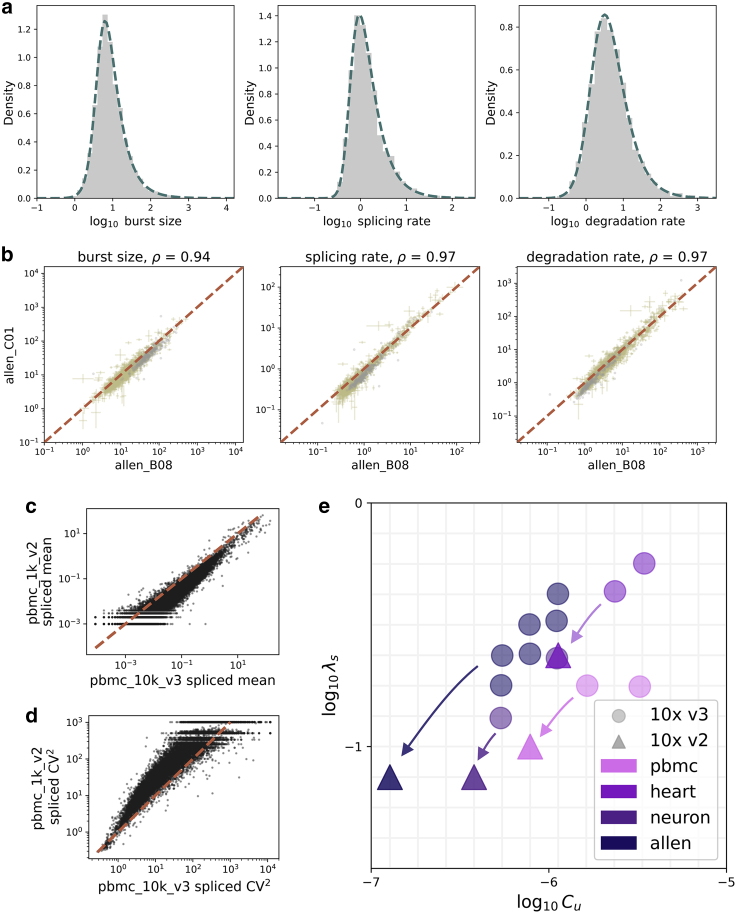
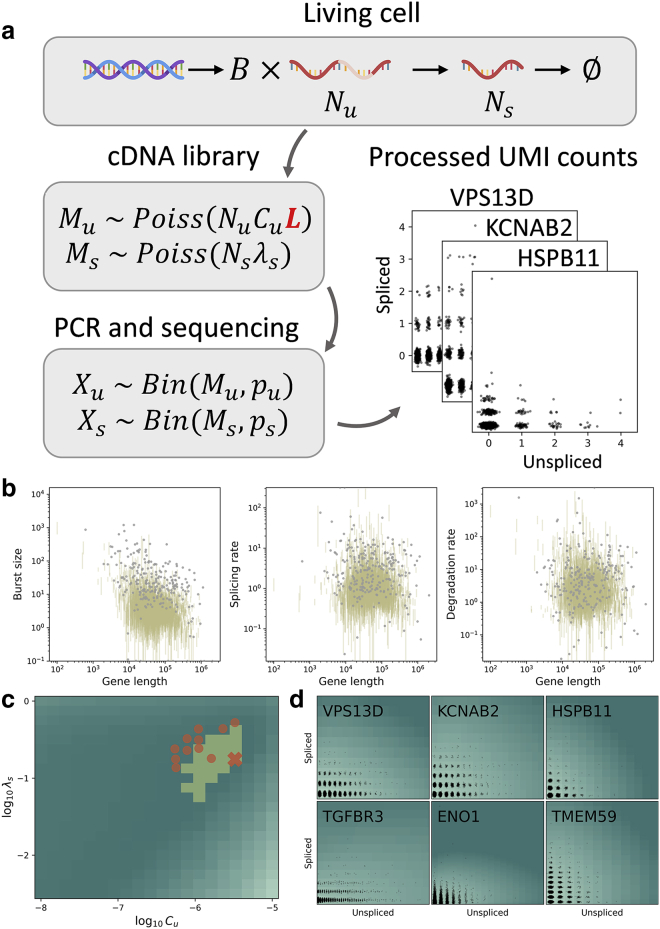
Single-cell RNA sequencing data can be modeled using Markov chains to yield genome-wide insights into transcriptional physics. However, quantitative inference with such data requires careful assessment of noise sources. We find that long pre-mRNA transcripts are over-represented in sequencing data. To explain this trend, we propose a length-based model of capture bias, which may produce false-positive observations. We solve this model and use it to find concordant parameter trends as well as systematic, mechanistically interpretable technical and biological differences in paired data sets.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


