{"title":"促进机器学习引导蛋白质工程与智能库设计和大规模并行分析","authors":"Hoi Yee Chu, Alan S. L. Wong","doi":"10.1002/ggn2.202100038","DOIUrl":null,"url":null,"abstract":"<p>Protein design plays an important role in recent medical advances from antibody therapy to vaccine design. Typically, exhaustive mutational screens or directed evolution experiments are used for the identification of the best design or for improvements to the wild-type variant. Even with a high-throughput screening on pooled libraries and Next-Generation Sequencing to boost the scale of read-outs, surveying all the variants with combinatorial mutations for their empirical fitness scores is still of magnitudes beyond the capacity of existing experimental settings. To tackle this challenge, in-silico approaches using machine learning to predict the fitness of novel variants based on a subset of empirical measurements are now employed. These machine learning models turn out to be useful in many cases, with the premise that the experimentally determined fitness scores and the amino-acid descriptors of the models are informative. The machine learning models can guide the search for the highest fitness variants, resolve complex epistatic relationships, and highlight bio-physical rules for protein folding. Using machine learning-guided approaches, researchers can build more focused libraries, thus relieving themselves from labor-intensive screens and fast-tracking the optimization process. Here, we describe the current advances in massive-scale variant screens, and how machine learning and mutagenesis strategies can be integrated to accelerate protein engineering. More specifically, we examine strategies to make screens more economical, informative, and effective in discovery of useful variants.</p>","PeriodicalId":72071,"journal":{"name":"Advanced genetics (Hoboken, N.J.)","volume":"2 4","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2021-12-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9744531/pdf/","citationCount":"2","resultStr":"{\"title\":\"Facilitating Machine Learning-Guided Protein Engineering with Smart Library Design and Massively Parallel Assays\",\"authors\":\"Hoi Yee Chu, Alan S. L. Wong\",\"doi\":\"10.1002/ggn2.202100038\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Protein design plays an important role in recent medical advances from antibody therapy to vaccine design. Typically, exhaustive mutational screens or directed evolution experiments are used for the identification of the best design or for improvements to the wild-type variant. Even with a high-throughput screening on pooled libraries and Next-Generation Sequencing to boost the scale of read-outs, surveying all the variants with combinatorial mutations for their empirical fitness scores is still of magnitudes beyond the capacity of existing experimental settings. To tackle this challenge, in-silico approaches using machine learning to predict the fitness of novel variants based on a subset of empirical measurements are now employed. These machine learning models turn out to be useful in many cases, with the premise that the experimentally determined fitness scores and the amino-acid descriptors of the models are informative. The machine learning models can guide the search for the highest fitness variants, resolve complex epistatic relationships, and highlight bio-physical rules for protein folding. Using machine learning-guided approaches, researchers can build more focused libraries, thus relieving themselves from labor-intensive screens and fast-tracking the optimization process. Here, we describe the current advances in massive-scale variant screens, and how machine learning and mutagenesis strategies can be integrated to accelerate protein engineering. More specifically, we examine strategies to make screens more economical, informative, and effective in discovery of useful variants.</p>\",\"PeriodicalId\":72071,\"journal\":{\"name\":\"Advanced genetics (Hoboken, N.J.)\",\"volume\":\"2 4\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-12-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9744531/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advanced genetics (Hoboken, N.J.)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ggn2.202100038\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced genetics (Hoboken, N.J.)","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ggn2.202100038","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Facilitating Machine Learning-Guided Protein Engineering with Smart Library Design and Massively Parallel Assays
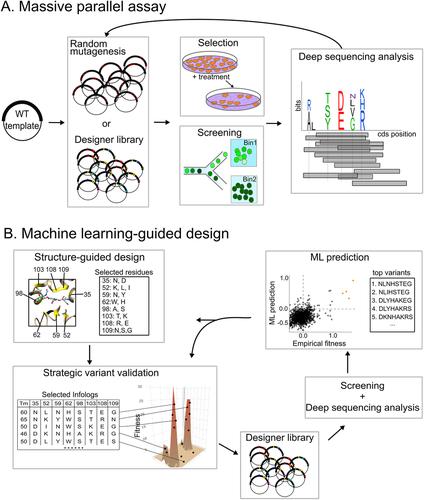
Protein design plays an important role in recent medical advances from antibody therapy to vaccine design. Typically, exhaustive mutational screens or directed evolution experiments are used for the identification of the best design or for improvements to the wild-type variant. Even with a high-throughput screening on pooled libraries and Next-Generation Sequencing to boost the scale of read-outs, surveying all the variants with combinatorial mutations for their empirical fitness scores is still of magnitudes beyond the capacity of existing experimental settings. To tackle this challenge, in-silico approaches using machine learning to predict the fitness of novel variants based on a subset of empirical measurements are now employed. These machine learning models turn out to be useful in many cases, with the premise that the experimentally determined fitness scores and the amino-acid descriptors of the models are informative. The machine learning models can guide the search for the highest fitness variants, resolve complex epistatic relationships, and highlight bio-physical rules for protein folding. Using machine learning-guided approaches, researchers can build more focused libraries, thus relieving themselves from labor-intensive screens and fast-tracking the optimization process. Here, we describe the current advances in massive-scale variant screens, and how machine learning and mutagenesis strategies can be integrated to accelerate protein engineering. More specifically, we examine strategies to make screens more economical, informative, and effective in discovery of useful variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


