{"title":"Dirichlet过程混合物中变量选择的快速近似推断,并在泛癌症蛋白质组学中的应用。","authors":"Oliver M Crook, Laurent Gatto, Paul D W Kirk","doi":"10.1515/sagmb-2018-0065","DOIUrl":null,"url":null,"abstract":"<p><p>The Dirichlet Process (DP) mixture model has become a popular choice for model-based clustering, largely because it allows the number of clusters to be inferred. The sequential updating and greedy search (SUGS) algorithm (Wang & Dunson, 2011) was proposed as a fast method for performing approximate Bayesian inference in DP mixture models, by posing clustering as a Bayesian model selection (BMS) problem and avoiding the use of computationally costly Markov chain Monte Carlo methods. Here we consider how this approach may be extended to permit variable selection for clustering, and also demonstrate the benefits of Bayesian model averaging (BMA) in place of BMS. Through an array of simulation examples and well-studied examples from cancer transcriptomics, we show that our method performs competitively with the current state-of-the-art, while also offering computational benefits. We apply our approach to reverse-phase protein array (RPPA) data from The Cancer Genome Atlas (TCGA) in order to perform a pan-cancer proteomic characterisation of 5157 tumour samples. We have implemented our approach, together with the original SUGS algorithm, in an open-source R package named sugsvarsel, which accelerates analysis by performing intensive computations in C++ and provides automated parallel processing. The R package is freely available from: https://github.com/ococrook/sugsvarsel.</p>","PeriodicalId":49477,"journal":{"name":"Statistical Applications in Genetics and Molecular Biology","volume":"18 6","pages":""},"PeriodicalIF":0.9000,"publicationDate":"2019-12-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7614016/pdf/","citationCount":"0","resultStr":"{\"title\":\"Fast approximate inference for variable selection in Dirichlet process mixtures, with an application to pan-cancer proteomics.\",\"authors\":\"Oliver M Crook, Laurent Gatto, Paul D W Kirk\",\"doi\":\"10.1515/sagmb-2018-0065\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The Dirichlet Process (DP) mixture model has become a popular choice for model-based clustering, largely because it allows the number of clusters to be inferred. The sequential updating and greedy search (SUGS) algorithm (Wang & Dunson, 2011) was proposed as a fast method for performing approximate Bayesian inference in DP mixture models, by posing clustering as a Bayesian model selection (BMS) problem and avoiding the use of computationally costly Markov chain Monte Carlo methods. Here we consider how this approach may be extended to permit variable selection for clustering, and also demonstrate the benefits of Bayesian model averaging (BMA) in place of BMS. Through an array of simulation examples and well-studied examples from cancer transcriptomics, we show that our method performs competitively with the current state-of-the-art, while also offering computational benefits. We apply our approach to reverse-phase protein array (RPPA) data from The Cancer Genome Atlas (TCGA) in order to perform a pan-cancer proteomic characterisation of 5157 tumour samples. We have implemented our approach, together with the original SUGS algorithm, in an open-source R package named sugsvarsel, which accelerates analysis by performing intensive computations in C++ and provides automated parallel processing. The R package is freely available from: https://github.com/ococrook/sugsvarsel.</p>\",\"PeriodicalId\":49477,\"journal\":{\"name\":\"Statistical Applications in Genetics and Molecular Biology\",\"volume\":\"18 6\",\"pages\":\"\"},\"PeriodicalIF\":0.9000,\"publicationDate\":\"2019-12-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7614016/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Statistical Applications in Genetics and Molecular Biology\",\"FirstCategoryId\":\"100\",\"ListUrlMain\":\"https://doi.org/10.1515/sagmb-2018-0065\",\"RegionNum\":4,\"RegionCategory\":\"数学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Mathematics\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Statistical Applications in Genetics and Molecular Biology","FirstCategoryId":"100","ListUrlMain":"https://doi.org/10.1515/sagmb-2018-0065","RegionNum":4,"RegionCategory":"数学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Mathematics","Score":null,"Total":0}
Fast approximate inference for variable selection in Dirichlet process mixtures, with an application to pan-cancer proteomics.
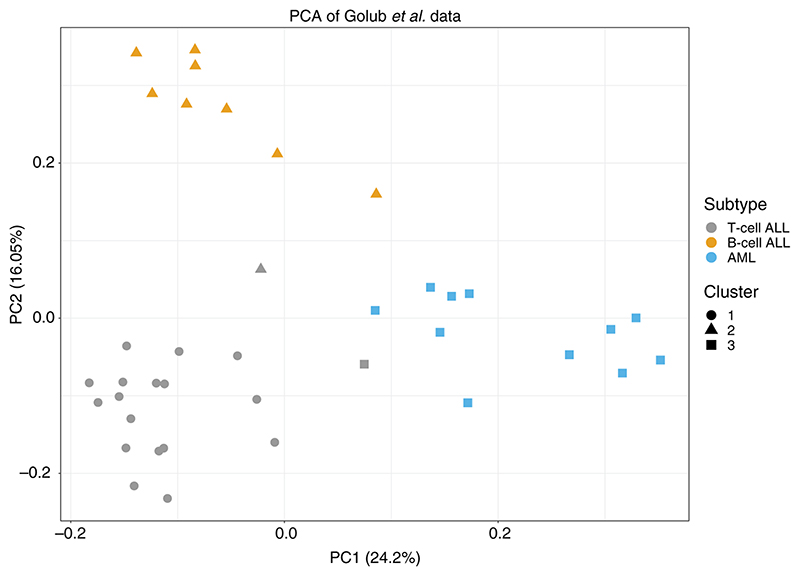
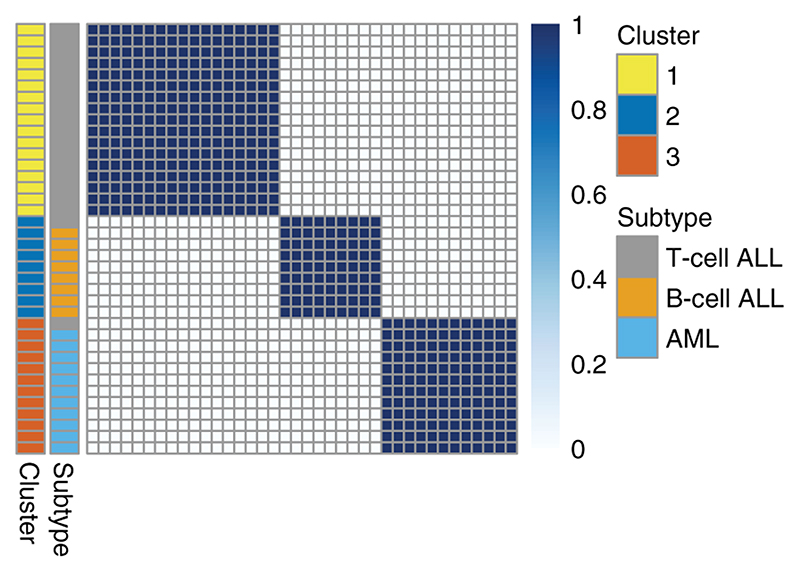
The Dirichlet Process (DP) mixture model has become a popular choice for model-based clustering, largely because it allows the number of clusters to be inferred. The sequential updating and greedy search (SUGS) algorithm (Wang & Dunson, 2011) was proposed as a fast method for performing approximate Bayesian inference in DP mixture models, by posing clustering as a Bayesian model selection (BMS) problem and avoiding the use of computationally costly Markov chain Monte Carlo methods. Here we consider how this approach may be extended to permit variable selection for clustering, and also demonstrate the benefits of Bayesian model averaging (BMA) in place of BMS. Through an array of simulation examples and well-studied examples from cancer transcriptomics, we show that our method performs competitively with the current state-of-the-art, while also offering computational benefits. We apply our approach to reverse-phase protein array (RPPA) data from The Cancer Genome Atlas (TCGA) in order to perform a pan-cancer proteomic characterisation of 5157 tumour samples. We have implemented our approach, together with the original SUGS algorithm, in an open-source R package named sugsvarsel, which accelerates analysis by performing intensive computations in C++ and provides automated parallel processing. The R package is freely available from: https://github.com/ococrook/sugsvarsel.
期刊介绍:
Statistical Applications in Genetics and Molecular Biology seeks to publish significant research on the application of statistical ideas to problems arising from computational biology. The focus of the papers should be on the relevant statistical issues but should contain a succinct description of the relevant biological problem being considered. The range of topics is wide and will include topics such as linkage mapping, association studies, gene finding and sequence alignment, protein structure prediction, design and analysis of microarray data, molecular evolution and phylogenetic trees, DNA topology, and data base search strategies. Both original research and review articles will be warmly received.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


