Yongshan He, Xuan Dai, Yuanyuan Chen, Shiyong Huang
{"title":"基因组和表达数据的综合分析确定了预测结直肠癌预后和免疫反应的潜在标志物。","authors":"Yongshan He, Xuan Dai, Yuanyuan Chen, Shiyong Huang","doi":"10.1155/2022/1831211","DOIUrl":null,"url":null,"abstract":"<p><p>Colorectal cancer (CRC) is the most prevalent type of malignant tumor of the gastrointestinal tract. In the current study, we characterized the landscape of genomic alterations in CRC patients. Based on the results of whole-exome sequencing (WES), we identified 31 significantly mutated genes. Among them, several genes including TP53, KRAS, APC, PI3KCA, and BRAF were reported as significantly mutated genes in previous studies. In the current study, the most frequently mutated gene was TP53, which encodes tumor suppressor p53, affecting approximately 60% of CRC patients. In addition, we performed the expression profiles of significantly mutated genes between the normal group and tumor groups and identified 20 differentially expressed genes (DEGs); among them, CSMD3, DCHS2, LRP2, RYR2, and ZFHX4 were significantly negatively correlated with PFS. Moreover, consensus clustering analysis for CRC based on the expression of significantly somatic mutated genes was performed. In total, three subtypes of CRC were identified in CRC, including cluster1 (<i>n</i> = 453), cluster2 (<i>n</i> = 158), and cluster 3 (<i>n</i> = 9), based on expression level of significantly somatic mutated genes. Clinicopathological features analysis showed subtype C1 had the longest progression-free survival (PFS) with median time of 8.2 years, while subtypes C2 and C3 had 4.1 and 2.7 years of PFS, respectively. Moreover, we found three subtypes related to tumor infiltration depth, lymph node metastasis, and distant metastasis. Immune infiltration analysis showed the tumor infiltration levels of B cell native, T cell CD8+, T cell CD4+ memory activated, T cell gamma delta, NK cell resting, macrophage M0, macrophage M2, myeloid dendritic cell activated, mast cell activated, and mast cell resting significantly changed among the three groups, demonstrating the three subgroups classified by 22 somatically significantly mutated genes had a high capacity to differentiate patients with different immune statuses, which is helpful for the prediction of immunotherapy response of CRC patients. Our findings could provide novel potential predictive indicators for CRC prognosis and therapy targets for CRC immunotherapy.</p>","PeriodicalId":12778,"journal":{"name":"Genetics research","volume":"2022 ","pages":"1831211"},"PeriodicalIF":1.4000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9356844/pdf/","citationCount":"2","resultStr":"{\"title\":\"Comprehensive Analysis of Genomic and Expression Data Identified Potential Markers for Predicting Prognosis and Immune Response in CRC.\",\"authors\":\"Yongshan He, Xuan Dai, Yuanyuan Chen, Shiyong Huang\",\"doi\":\"10.1155/2022/1831211\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Colorectal cancer (CRC) is the most prevalent type of malignant tumor of the gastrointestinal tract. In the current study, we characterized the landscape of genomic alterations in CRC patients. Based on the results of whole-exome sequencing (WES), we identified 31 significantly mutated genes. Among them, several genes including TP53, KRAS, APC, PI3KCA, and BRAF were reported as significantly mutated genes in previous studies. In the current study, the most frequently mutated gene was TP53, which encodes tumor suppressor p53, affecting approximately 60% of CRC patients. In addition, we performed the expression profiles of significantly mutated genes between the normal group and tumor groups and identified 20 differentially expressed genes (DEGs); among them, CSMD3, DCHS2, LRP2, RYR2, and ZFHX4 were significantly negatively correlated with PFS. Moreover, consensus clustering analysis for CRC based on the expression of significantly somatic mutated genes was performed. In total, three subtypes of CRC were identified in CRC, including cluster1 (<i>n</i> = 453), cluster2 (<i>n</i> = 158), and cluster 3 (<i>n</i> = 9), based on expression level of significantly somatic mutated genes. Clinicopathological features analysis showed subtype C1 had the longest progression-free survival (PFS) with median time of 8.2 years, while subtypes C2 and C3 had 4.1 and 2.7 years of PFS, respectively. Moreover, we found three subtypes related to tumor infiltration depth, lymph node metastasis, and distant metastasis. Immune infiltration analysis showed the tumor infiltration levels of B cell native, T cell CD8+, T cell CD4+ memory activated, T cell gamma delta, NK cell resting, macrophage M0, macrophage M2, myeloid dendritic cell activated, mast cell activated, and mast cell resting significantly changed among the three groups, demonstrating the three subgroups classified by 22 somatically significantly mutated genes had a high capacity to differentiate patients with different immune statuses, which is helpful for the prediction of immunotherapy response of CRC patients. Our findings could provide novel potential predictive indicators for CRC prognosis and therapy targets for CRC immunotherapy.</p>\",\"PeriodicalId\":12778,\"journal\":{\"name\":\"Genetics research\",\"volume\":\"2022 \",\"pages\":\"1831211\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9356844/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1155/2022/1831211\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1155/2022/1831211","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Comprehensive Analysis of Genomic and Expression Data Identified Potential Markers for Predicting Prognosis and Immune Response in CRC.
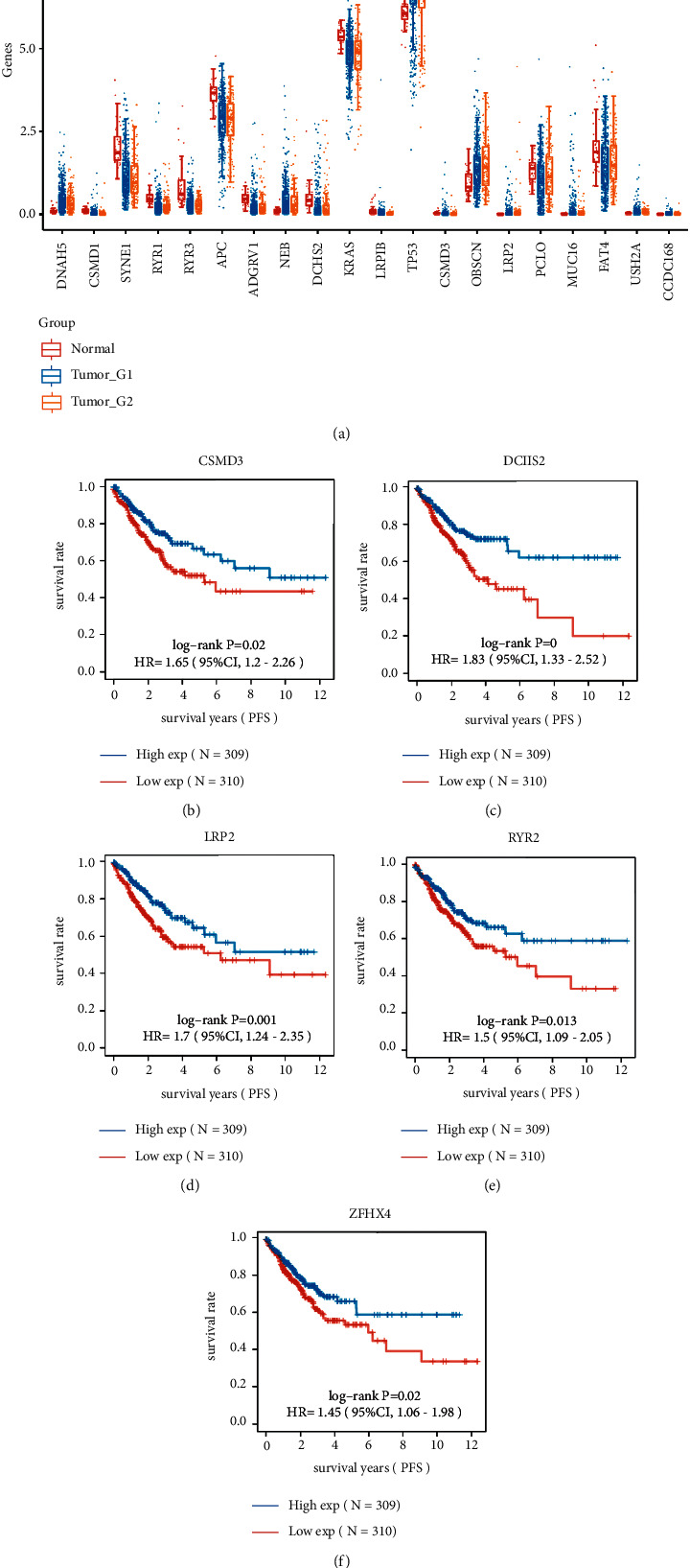
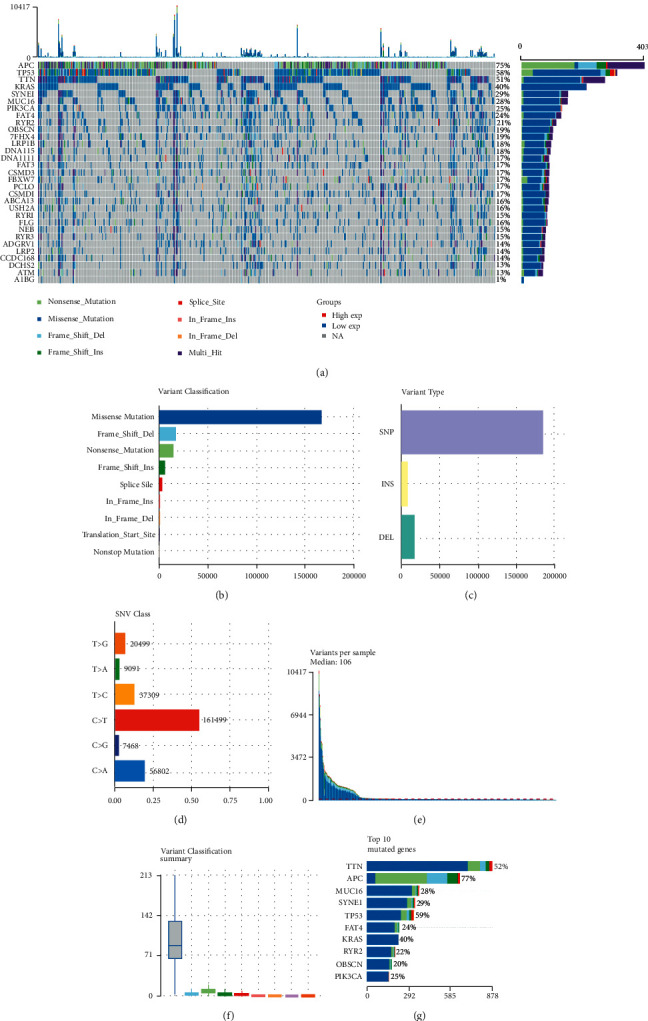
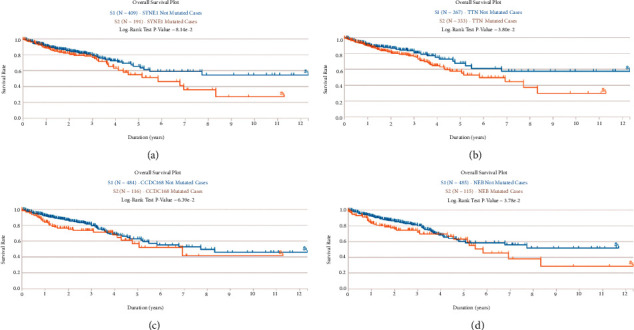
Colorectal cancer (CRC) is the most prevalent type of malignant tumor of the gastrointestinal tract. In the current study, we characterized the landscape of genomic alterations in CRC patients. Based on the results of whole-exome sequencing (WES), we identified 31 significantly mutated genes. Among them, several genes including TP53, KRAS, APC, PI3KCA, and BRAF were reported as significantly mutated genes in previous studies. In the current study, the most frequently mutated gene was TP53, which encodes tumor suppressor p53, affecting approximately 60% of CRC patients. In addition, we performed the expression profiles of significantly mutated genes between the normal group and tumor groups and identified 20 differentially expressed genes (DEGs); among them, CSMD3, DCHS2, LRP2, RYR2, and ZFHX4 were significantly negatively correlated with PFS. Moreover, consensus clustering analysis for CRC based on the expression of significantly somatic mutated genes was performed. In total, three subtypes of CRC were identified in CRC, including cluster1 (n = 453), cluster2 (n = 158), and cluster 3 (n = 9), based on expression level of significantly somatic mutated genes. Clinicopathological features analysis showed subtype C1 had the longest progression-free survival (PFS) with median time of 8.2 years, while subtypes C2 and C3 had 4.1 and 2.7 years of PFS, respectively. Moreover, we found three subtypes related to tumor infiltration depth, lymph node metastasis, and distant metastasis. Immune infiltration analysis showed the tumor infiltration levels of B cell native, T cell CD8+, T cell CD4+ memory activated, T cell gamma delta, NK cell resting, macrophage M0, macrophage M2, myeloid dendritic cell activated, mast cell activated, and mast cell resting significantly changed among the three groups, demonstrating the three subgroups classified by 22 somatically significantly mutated genes had a high capacity to differentiate patients with different immune statuses, which is helpful for the prediction of immunotherapy response of CRC patients. Our findings could provide novel potential predictive indicators for CRC prognosis and therapy targets for CRC immunotherapy.
期刊介绍:
Genetics Research is a key forum for original research on all aspects of human and animal genetics, reporting key findings on genomes, genes, mutations and molecular interactions, extending out to developmental, evolutionary, and population genetics as well as ethical, legal and social aspects. Our aim is to lead to a better understanding of genetic processes in health and disease. The journal focuses on the use of new technologies, such as next generation sequencing together with bioinformatics analysis, to produce increasingly detailed views of how genes function in tissues and how these genes perform, individually or collectively, in normal development and disease aetiology. The journal publishes original work, review articles, short papers, computational studies, and novel methods and techniques in research covering humans and well-established genetic organisms. Key subject areas include medical genetics, genomics, human evolutionary and population genetics, bioinformatics, genetics of complex traits, molecular and developmental genetics, Evo-Devo, quantitative and statistical genetics, behavioural genetics and environmental genetics. The breadth and quality of research make the journal an invaluable resource for medical geneticists, molecular biologists, bioinformaticians and researchers involved in genetic basis of diseases, evolutionary and developmental studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


