{"title":"ClusterDE:一种对双浸渍引起的假阳性膨胀具有鲁棒性的聚类后差异表达(DE)方法。","authors":"Dongyuan Song, Siqi Chen, Christy Lee, Kexin Li, Xinzhou Ge, Jingyi Jessica Li","doi":"10.1101/2023.07.21.550107","DOIUrl":null,"url":null,"abstract":"<p><p>Double dipping is a well-known pitfall in single-cell and spatial transcriptomics data analysis: after a clustering algorithm finds clusters as putative cell types or spatial domains, statistical tests are applied to the same data to identify differentially expressed (DE) genes as potential cell-type or spatial-domain markers. Because the genes that contribute to clustering are inherently likely to be identified as DE genes, double dipping can result in false-positive cell-type or spatial-domain markers, especially when clusters are spurious, leading to ambiguously defined cell types or spatial domains. To address this challenge, we propose ClusterDE, a statistical method designed to identify post-clustering DE genes as reliable markers of cell types and spatial domains, while controlling the false discovery rate (FDR) regardless of clustering quality. The core of ClusterDE involves generating synthetic null data as an <i>in silico</i> negative control that contains only one cell type or spatial domain, allowing for the detection and removal of spurious discoveries caused by double dipping. We demonstrate that ClusterDE controls the FDR and identifies canonical cell-type and spatial-domain markers as top DE genes, distinguishing them from housekeeping genes. ClusterDE's ability to discover reliable markers, or the absence of such markers, can be used to determine whether two ambiguous clusters should be merged. Additionally, ClusterDE is compatible with state-of-the-art analysis pipelines like Seurat and Scanpy.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/91/79/nihpp-2023.07.21.550107v1.PMC10401959.pdf","citationCount":"0","resultStr":"{\"title\":\"Synthetic control removes spurious discoveries from double dipping in single-cell and spatial transcriptomics data analyses.\",\"authors\":\"Dongyuan Song, Siqi Chen, Christy Lee, Kexin Li, Xinzhou Ge, Jingyi Jessica Li\",\"doi\":\"10.1101/2023.07.21.550107\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Double dipping is a well-known pitfall in single-cell and spatial transcriptomics data analysis: after a clustering algorithm finds clusters as putative cell types or spatial domains, statistical tests are applied to the same data to identify differentially expressed (DE) genes as potential cell-type or spatial-domain markers. Because the genes that contribute to clustering are inherently likely to be identified as DE genes, double dipping can result in false-positive cell-type or spatial-domain markers, especially when clusters are spurious, leading to ambiguously defined cell types or spatial domains. To address this challenge, we propose ClusterDE, a statistical method designed to identify post-clustering DE genes as reliable markers of cell types and spatial domains, while controlling the false discovery rate (FDR) regardless of clustering quality. The core of ClusterDE involves generating synthetic null data as an <i>in silico</i> negative control that contains only one cell type or spatial domain, allowing for the detection and removal of spurious discoveries caused by double dipping. We demonstrate that ClusterDE controls the FDR and identifies canonical cell-type and spatial-domain markers as top DE genes, distinguishing them from housekeeping genes. ClusterDE's ability to discover reliable markers, or the absence of such markers, can be used to determine whether two ambiguous clusters should be merged. Additionally, ClusterDE is compatible with state-of-the-art analysis pipelines like Seurat and Scanpy.</p>\",\"PeriodicalId\":72407,\"journal\":{\"name\":\"bioRxiv : the preprint server for biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-12-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/91/79/nihpp-2023.07.21.550107v1.PMC10401959.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"bioRxiv : the preprint server for biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.07.21.550107\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.07.21.550107","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Synthetic control removes spurious discoveries from double dipping in single-cell and spatial transcriptomics data analyses.
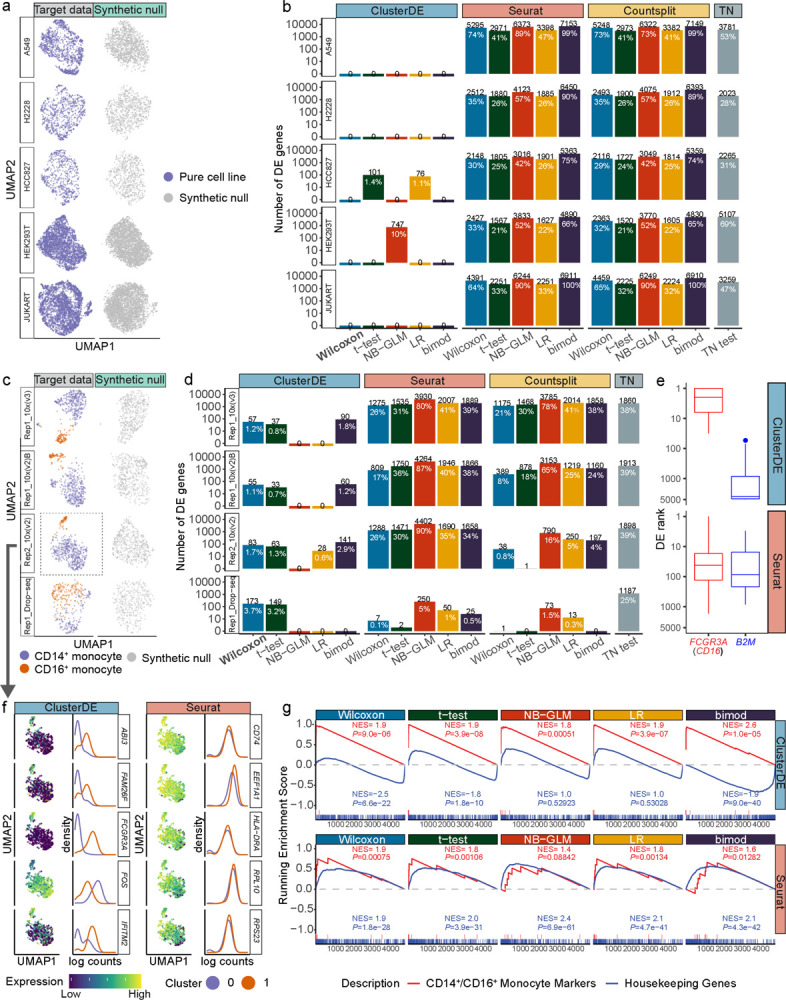
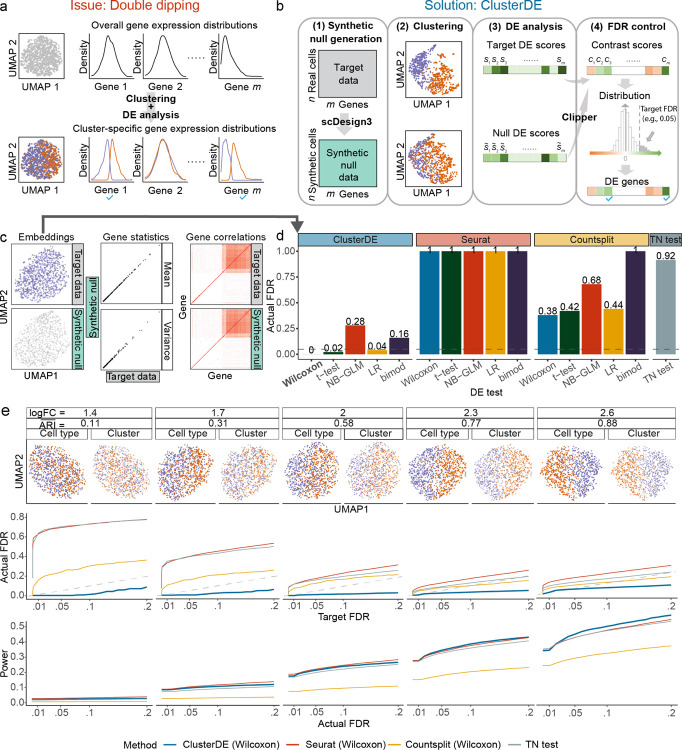
Double dipping is a well-known pitfall in single-cell and spatial transcriptomics data analysis: after a clustering algorithm finds clusters as putative cell types or spatial domains, statistical tests are applied to the same data to identify differentially expressed (DE) genes as potential cell-type or spatial-domain markers. Because the genes that contribute to clustering are inherently likely to be identified as DE genes, double dipping can result in false-positive cell-type or spatial-domain markers, especially when clusters are spurious, leading to ambiguously defined cell types or spatial domains. To address this challenge, we propose ClusterDE, a statistical method designed to identify post-clustering DE genes as reliable markers of cell types and spatial domains, while controlling the false discovery rate (FDR) regardless of clustering quality. The core of ClusterDE involves generating synthetic null data as an in silico negative control that contains only one cell type or spatial domain, allowing for the detection and removal of spurious discoveries caused by double dipping. We demonstrate that ClusterDE controls the FDR and identifies canonical cell-type and spatial-domain markers as top DE genes, distinguishing them from housekeeping genes. ClusterDE's ability to discover reliable markers, or the absence of such markers, can be used to determine whether two ambiguous clusters should be merged. Additionally, ClusterDE is compatible with state-of-the-art analysis pipelines like Seurat and Scanpy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


