{"title":"弗里德里希共济失调干预措施评估的临床证据:一项系统综述。","authors":"Paridhi Jain, Lohit Badgujar, Jelle Spoorendonk, Katharina Buesch","doi":"10.1177/26330040221139872","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>The rare inherited autosomal recessive disease Friedreich ataxia (FA) causes progressive neurodegenerative changes and disability in patients. A systematic literature review (SLR) was carried out to understand and summarize the published efficacy and safety of therapeutic interventions in this disease.</p><p><strong>Methods: </strong>Database searches were carried out in MEDLINE, Embase, and Cochrane by two independent reviewers. In addition, trial registries and conference proceedings were hand-searched.</p><p><strong>Results: </strong>Thirty-two publications were deemed eligible according to PICOS criteria. Twenty-four publications detail randomized controlled trials. The most frequently identified therapeutic intervention was idebenone (<i>n</i> = 11), followed by recombinant erythropoietin (<i>n</i> = 6), omaveloxolone (<i>n</i> = 3), and amantadine hydrochloride (<i>n</i> = 2). Other therapeutic interventions were investigated in one publication: A0001, CoQ10, creatine, deferiprone, interferon-γ-1b, the L-carnitine levorotatory form of 5-hydroxytryptophan, luvadaxistat, resveratrol, RT001, and vatiquinone (EPI-743). These studies included patients from 8 to 73 years old, and disease duration varied from 4.7 to 19 years. Disease severity as per the mean GAA1 and GAA2 allele repeat length ranged from 350 to 930 and 620 to 987 nucleotides, respectively. Most frequently reported efficacy outcomes were the International Cooperative Ataxia Rating Scale (ICARS, <i>n</i> = 10), the Friedreich Ataxia Rating Scale (modified FARS and FARS-neuro, <i>n</i> = 12), the Scale for Assessment and Rating of Ataxia (SARA, <i>n</i> = 7), and the Activities of Daily Living scale (ADL, <i>n</i> = 8). Each of these assesses the severity of disability in FA patients. In many studies, patients with FA deteriorated according to these severity scales regardless of treatment, or inconclusive results were found. Generally, these therapeutic interventions were well-tolerated and safe. Serious adverse events were atrial fibrillation (<i>n</i> = 1), craniocerebral injury (<i>n</i> = 1), and ventricular tachycardia (<i>n</i> = 1).</p><p><strong>Conclusion: </strong>Identified literature showed a considerable unmet need for therapeutic interventions that halt or slow the deteriorating nature of FA. Novel efficacious drugs should be investigated that aim to improve symptoms or slow disease progression.</p>","PeriodicalId":75218,"journal":{"name":"Therapeutic advances in rare disease","volume":"3 ","pages":"26330040221139872"},"PeriodicalIF":0.0000,"publicationDate":"2022-11-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6e/67/10.1177_26330040221139872.PMC10032438.pdf","citationCount":"3","resultStr":"{\"title\":\"Clinical evidence of interventions assessed in Friedreich ataxia: a systematic review.\",\"authors\":\"Paridhi Jain, Lohit Badgujar, Jelle Spoorendonk, Katharina Buesch\",\"doi\":\"10.1177/26330040221139872\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objectives: </strong>The rare inherited autosomal recessive disease Friedreich ataxia (FA) causes progressive neurodegenerative changes and disability in patients. A systematic literature review (SLR) was carried out to understand and summarize the published efficacy and safety of therapeutic interventions in this disease.</p><p><strong>Methods: </strong>Database searches were carried out in MEDLINE, Embase, and Cochrane by two independent reviewers. In addition, trial registries and conference proceedings were hand-searched.</p><p><strong>Results: </strong>Thirty-two publications were deemed eligible according to PICOS criteria. Twenty-four publications detail randomized controlled trials. The most frequently identified therapeutic intervention was idebenone (<i>n</i> = 11), followed by recombinant erythropoietin (<i>n</i> = 6), omaveloxolone (<i>n</i> = 3), and amantadine hydrochloride (<i>n</i> = 2). Other therapeutic interventions were investigated in one publication: A0001, CoQ10, creatine, deferiprone, interferon-γ-1b, the L-carnitine levorotatory form of 5-hydroxytryptophan, luvadaxistat, resveratrol, RT001, and vatiquinone (EPI-743). These studies included patients from 8 to 73 years old, and disease duration varied from 4.7 to 19 years. Disease severity as per the mean GAA1 and GAA2 allele repeat length ranged from 350 to 930 and 620 to 987 nucleotides, respectively. Most frequently reported efficacy outcomes were the International Cooperative Ataxia Rating Scale (ICARS, <i>n</i> = 10), the Friedreich Ataxia Rating Scale (modified FARS and FARS-neuro, <i>n</i> = 12), the Scale for Assessment and Rating of Ataxia (SARA, <i>n</i> = 7), and the Activities of Daily Living scale (ADL, <i>n</i> = 8). Each of these assesses the severity of disability in FA patients. In many studies, patients with FA deteriorated according to these severity scales regardless of treatment, or inconclusive results were found. Generally, these therapeutic interventions were well-tolerated and safe. Serious adverse events were atrial fibrillation (<i>n</i> = 1), craniocerebral injury (<i>n</i> = 1), and ventricular tachycardia (<i>n</i> = 1).</p><p><strong>Conclusion: </strong>Identified literature showed a considerable unmet need for therapeutic interventions that halt or slow the deteriorating nature of FA. Novel efficacious drugs should be investigated that aim to improve symptoms or slow disease progression.</p>\",\"PeriodicalId\":75218,\"journal\":{\"name\":\"Therapeutic advances in rare disease\",\"volume\":\"3 \",\"pages\":\"26330040221139872\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-11-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6e/67/10.1177_26330040221139872.PMC10032438.pdf\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Therapeutic advances in rare disease\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/26330040221139872\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Therapeutic advances in rare disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/26330040221139872","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Clinical evidence of interventions assessed in Friedreich ataxia: a systematic review.
Objectives: The rare inherited autosomal recessive disease Friedreich ataxia (FA) causes progressive neurodegenerative changes and disability in patients. A systematic literature review (SLR) was carried out to understand and summarize the published efficacy and safety of therapeutic interventions in this disease.
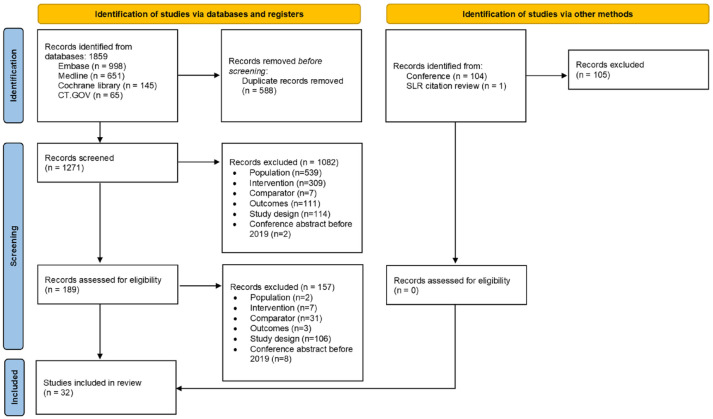
Methods: Database searches were carried out in MEDLINE, Embase, and Cochrane by two independent reviewers. In addition, trial registries and conference proceedings were hand-searched.
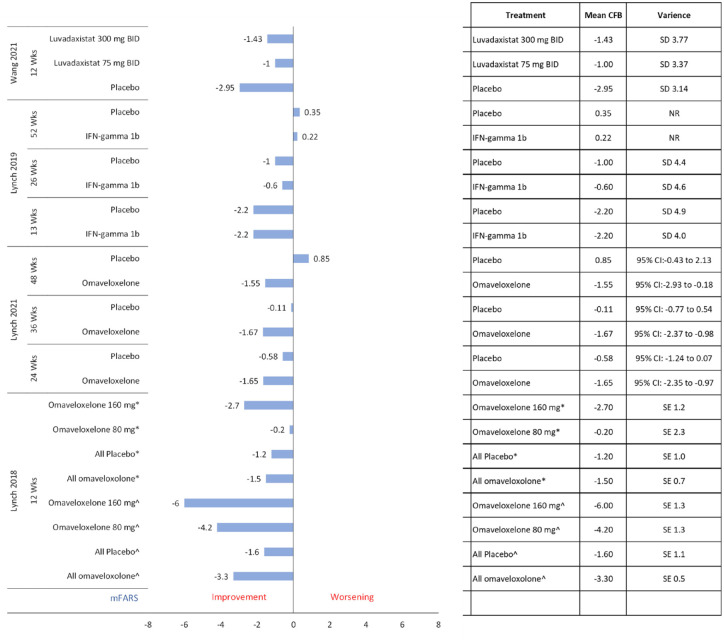
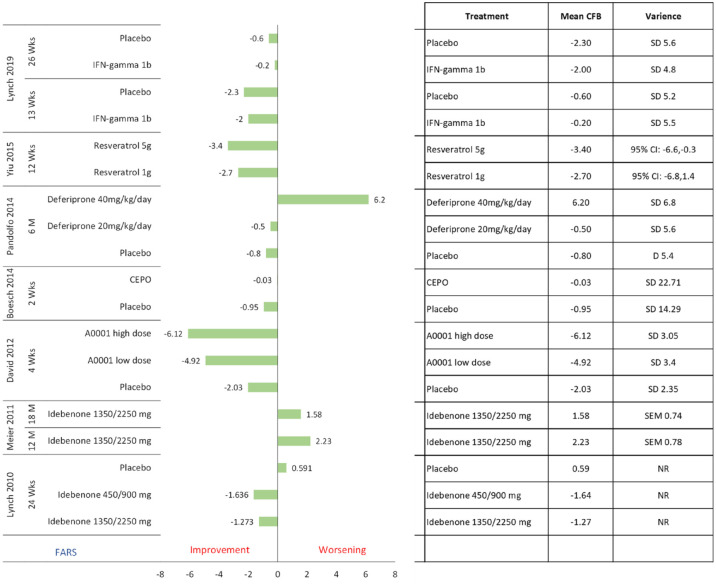
Results: Thirty-two publications were deemed eligible according to PICOS criteria. Twenty-four publications detail randomized controlled trials. The most frequently identified therapeutic intervention was idebenone (n = 11), followed by recombinant erythropoietin (n = 6), omaveloxolone (n = 3), and amantadine hydrochloride (n = 2). Other therapeutic interventions were investigated in one publication: A0001, CoQ10, creatine, deferiprone, interferon-γ-1b, the L-carnitine levorotatory form of 5-hydroxytryptophan, luvadaxistat, resveratrol, RT001, and vatiquinone (EPI-743). These studies included patients from 8 to 73 years old, and disease duration varied from 4.7 to 19 years. Disease severity as per the mean GAA1 and GAA2 allele repeat length ranged from 350 to 930 and 620 to 987 nucleotides, respectively. Most frequently reported efficacy outcomes were the International Cooperative Ataxia Rating Scale (ICARS, n = 10), the Friedreich Ataxia Rating Scale (modified FARS and FARS-neuro, n = 12), the Scale for Assessment and Rating of Ataxia (SARA, n = 7), and the Activities of Daily Living scale (ADL, n = 8). Each of these assesses the severity of disability in FA patients. In many studies, patients with FA deteriorated according to these severity scales regardless of treatment, or inconclusive results were found. Generally, these therapeutic interventions were well-tolerated and safe. Serious adverse events were atrial fibrillation (n = 1), craniocerebral injury (n = 1), and ventricular tachycardia (n = 1).
Conclusion: Identified literature showed a considerable unmet need for therapeutic interventions that halt or slow the deteriorating nature of FA. Novel efficacious drugs should be investigated that aim to improve symptoms or slow disease progression.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


