Sophie Wharrie, Zhiyu Yang, Vishnu Raj, Remo Monti, Rahul Gupta, Ying Wang, Alicia Martin, Luke J O'Connor, Samuel Kaski, Pekka Marttinen, Pier Francesco Palamara, Christoph Lippert, Andrea Ganna
{"title":"HAPNEST:高效、大规模地生成和评估基因型和表型的合成数据集。","authors":"Sophie Wharrie, Zhiyu Yang, Vishnu Raj, Remo Monti, Rahul Gupta, Ying Wang, Alicia Martin, Luke J O'Connor, Samuel Kaski, Pekka Marttinen, Pier Francesco Palamara, Christoph Lippert, Andrea Ganna","doi":"10.1093/bioinformatics/btad535","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Existing methods for simulating synthetic genotype and phenotype datasets have limited scalability, constraining their usability for large-scale analyses. Moreover, a systematic approach for evaluating synthetic data quality and a benchmark synthetic dataset for developing and evaluating methods for polygenic risk scores are lacking.</p><p><strong>Results: </strong>We present HAPNEST, a novel approach for efficiently generating diverse individual-level genotypic and phenotypic data. In comparison to alternative methods, HAPNEST shows faster computational speed and a lower degree of relatedness with reference panels, while generating datasets that preserve key statistical properties of real data. These desirable synthetic data properties enabled us to generate 6.8 million common variants and nine phenotypes with varying degrees of heritability and polygenicity across 1 million individuals. We demonstrate how HAPNEST can facilitate biobank-scale analyses through the comparison of seven methods to generate polygenic risk scoring across multiple ancestry groups and different genetic architectures.</p><p><strong>Availability and implementation: </strong>A synthetic dataset of 1 008 000 individuals and nine traits for 6.8 million common variants is available at https://www.ebi.ac.uk/biostudies/studies/S-BSST936. The HAPNEST software for generating synthetic datasets is available as Docker/Singularity containers and open source Julia and C code at https://github.com/intervene-EU-H2020/synthetic_data.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10493177/pdf/","citationCount":"0","resultStr":"{\"title\":\"HAPNEST: efficient, large-scale generation and evaluation of synthetic datasets for genotypes and phenotypes.\",\"authors\":\"Sophie Wharrie, Zhiyu Yang, Vishnu Raj, Remo Monti, Rahul Gupta, Ying Wang, Alicia Martin, Luke J O'Connor, Samuel Kaski, Pekka Marttinen, Pier Francesco Palamara, Christoph Lippert, Andrea Ganna\",\"doi\":\"10.1093/bioinformatics/btad535\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Existing methods for simulating synthetic genotype and phenotype datasets have limited scalability, constraining their usability for large-scale analyses. Moreover, a systematic approach for evaluating synthetic data quality and a benchmark synthetic dataset for developing and evaluating methods for polygenic risk scores are lacking.</p><p><strong>Results: </strong>We present HAPNEST, a novel approach for efficiently generating diverse individual-level genotypic and phenotypic data. In comparison to alternative methods, HAPNEST shows faster computational speed and a lower degree of relatedness with reference panels, while generating datasets that preserve key statistical properties of real data. These desirable synthetic data properties enabled us to generate 6.8 million common variants and nine phenotypes with varying degrees of heritability and polygenicity across 1 million individuals. We demonstrate how HAPNEST can facilitate biobank-scale analyses through the comparison of seven methods to generate polygenic risk scoring across multiple ancestry groups and different genetic architectures.</p><p><strong>Availability and implementation: </strong>A synthetic dataset of 1 008 000 individuals and nine traits for 6.8 million common variants is available at https://www.ebi.ac.uk/biostudies/studies/S-BSST936. The HAPNEST software for generating synthetic datasets is available as Docker/Singularity containers and open source Julia and C code at https://github.com/intervene-EU-H2020/synthetic_data.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 9\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10493177/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad535\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad535","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
HAPNEST: efficient, large-scale generation and evaluation of synthetic datasets for genotypes and phenotypes.
Motivation: Existing methods for simulating synthetic genotype and phenotype datasets have limited scalability, constraining their usability for large-scale analyses. Moreover, a systematic approach for evaluating synthetic data quality and a benchmark synthetic dataset for developing and evaluating methods for polygenic risk scores are lacking.
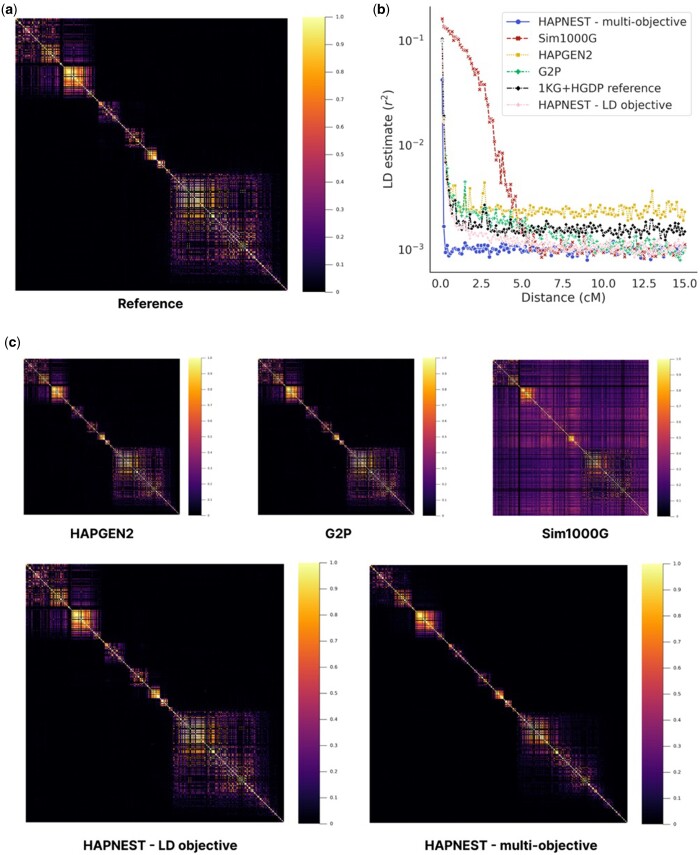
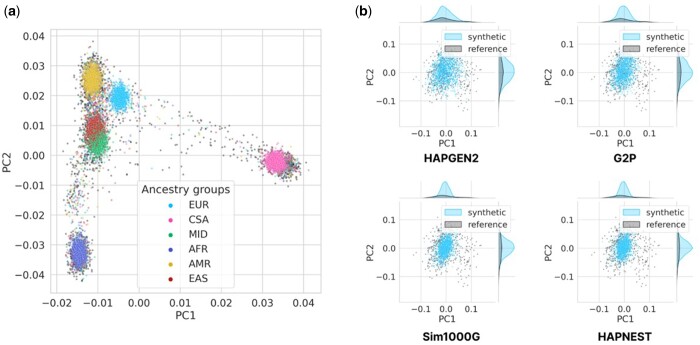
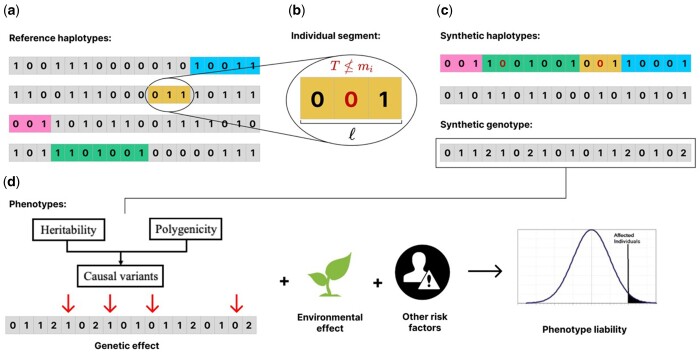
Results: We present HAPNEST, a novel approach for efficiently generating diverse individual-level genotypic and phenotypic data. In comparison to alternative methods, HAPNEST shows faster computational speed and a lower degree of relatedness with reference panels, while generating datasets that preserve key statistical properties of real data. These desirable synthetic data properties enabled us to generate 6.8 million common variants and nine phenotypes with varying degrees of heritability and polygenicity across 1 million individuals. We demonstrate how HAPNEST can facilitate biobank-scale analyses through the comparison of seven methods to generate polygenic risk scoring across multiple ancestry groups and different genetic architectures.
Availability and implementation: A synthetic dataset of 1 008 000 individuals and nine traits for 6.8 million common variants is available at https://www.ebi.ac.uk/biostudies/studies/S-BSST936. The HAPNEST software for generating synthetic datasets is available as Docker/Singularity containers and open source Julia and C code at https://github.com/intervene-EU-H2020/synthetic_data.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


