{"title":"DEP2:用于定量蛋白质组学数据的升级综合分析工具包。","authors":"Zhenhuan Feng, Peiyang Fang, Hui Zheng, Xiaofei Zhang","doi":"10.1093/bioinformatics/btad526","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>Mass spectrometry (MS)-based proteomics has become the most powerful approach to study the proteome of given biological and clinical samples. Advancements in sample preparation and MS detection have extended the application of proteomics but have also brought new demands on data analysis. Appropriate proteomics data analysis workflow mainly requires quality control, hypothesis testing, functional mining, and visualization. Although there are numerous tools for each process, an efficient and universal tandem analysis toolkit to obtain a quick overall view of various proteomics data is still urgently needed. Here, we present DEP2, an updated version of DEP we previously established, for proteomics data analysis. We amended the analysis workflow by incorporating alternative approaches to accommodate diverse proteomics data, introducing peptide-protein summarization and coupling biological function exploration. In summary, DEP2 is a well-rounded toolkit designed for protein- and peptide-level quantitative proteomics data. It features a more flexible differential analysis workflow and includes a user-friendly Shiny application to facilitate data analysis.</p><p><strong>Availability and implementation: </strong>DEP2 is available at https://github.com/mildpiggy/DEP2, released under the MIT license. For further information and usage details, please refer to the package website at https://mildpiggy.github.io/DEP2/.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 8","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10466079/pdf/","citationCount":"0","resultStr":"{\"title\":\"DEP2: an upgraded comprehensive analysis toolkit for quantitative proteomics data.\",\"authors\":\"Zhenhuan Feng, Peiyang Fang, Hui Zheng, Xiaofei Zhang\",\"doi\":\"10.1093/bioinformatics/btad526\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Summary: </strong>Mass spectrometry (MS)-based proteomics has become the most powerful approach to study the proteome of given biological and clinical samples. Advancements in sample preparation and MS detection have extended the application of proteomics but have also brought new demands on data analysis. Appropriate proteomics data analysis workflow mainly requires quality control, hypothesis testing, functional mining, and visualization. Although there are numerous tools for each process, an efficient and universal tandem analysis toolkit to obtain a quick overall view of various proteomics data is still urgently needed. Here, we present DEP2, an updated version of DEP we previously established, for proteomics data analysis. We amended the analysis workflow by incorporating alternative approaches to accommodate diverse proteomics data, introducing peptide-protein summarization and coupling biological function exploration. In summary, DEP2 is a well-rounded toolkit designed for protein- and peptide-level quantitative proteomics data. It features a more flexible differential analysis workflow and includes a user-friendly Shiny application to facilitate data analysis.</p><p><strong>Availability and implementation: </strong>DEP2 is available at https://github.com/mildpiggy/DEP2, released under the MIT license. For further information and usage details, please refer to the package website at https://mildpiggy.github.io/DEP2/.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 8\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10466079/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad526\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad526","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
DEP2: an upgraded comprehensive analysis toolkit for quantitative proteomics data.
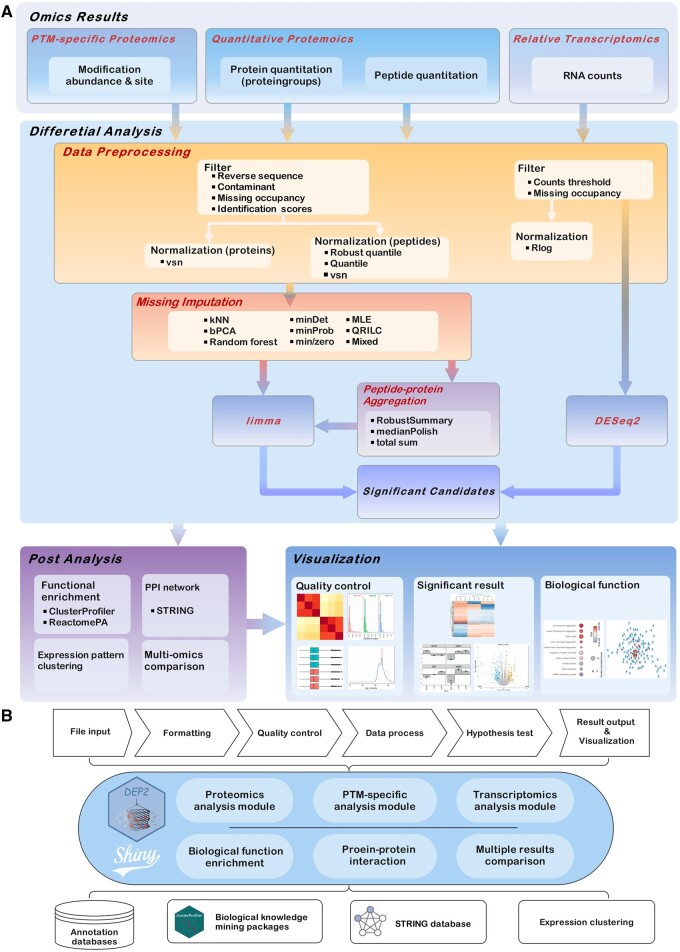
Summary: Mass spectrometry (MS)-based proteomics has become the most powerful approach to study the proteome of given biological and clinical samples. Advancements in sample preparation and MS detection have extended the application of proteomics but have also brought new demands on data analysis. Appropriate proteomics data analysis workflow mainly requires quality control, hypothesis testing, functional mining, and visualization. Although there are numerous tools for each process, an efficient and universal tandem analysis toolkit to obtain a quick overall view of various proteomics data is still urgently needed. Here, we present DEP2, an updated version of DEP we previously established, for proteomics data analysis. We amended the analysis workflow by incorporating alternative approaches to accommodate diverse proteomics data, introducing peptide-protein summarization and coupling biological function exploration. In summary, DEP2 is a well-rounded toolkit designed for protein- and peptide-level quantitative proteomics data. It features a more flexible differential analysis workflow and includes a user-friendly Shiny application to facilitate data analysis.
Availability and implementation: DEP2 is available at https://github.com/mildpiggy/DEP2, released under the MIT license. For further information and usage details, please refer to the package website at https://mildpiggy.github.io/DEP2/.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


