Hui Yu, Limin Jiang, Chung-I Li, Scott Ness, Sara G M Piccirillo, Yan Guo
{"title":"体细胞突变效应扩散到microRNA失调。","authors":"Hui Yu, Limin Jiang, Chung-I Li, Scott Ness, Sara G M Piccirillo, Yan Guo","doi":"10.1093/bioinformatics/btad520","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>As an important player in transcriptome regulation, microRNAs may effectively diffuse somatic mutation impacts to broad cellular processes and ultimately manifest disease and dictate prognosis. Previous studies that tried to correlate mutation with gene expression dysregulation neglected to adjust for the disparate multitudes of false positives associated with unequal sample sizes and uneven class balancing scenarios.</p><p><strong>Results: </strong>To properly address this issue, we developed a statistical framework to rigorously assess the extent of mutation impact on microRNAs in relation to a permutation-based null distribution of a matching sample structure. Carrying out the framework in a pan-cancer study, we ascertained 9008 protein-coding genes with statistically significant mutation impacts on miRNAs. Of these, the collective miRNA expression for 83 genes showed significant prognostic power in nine cancer types. For example, in lower-grade glioma, 10 genes' mutations broadly impacted miRNAs, all of which showed prognostic value with the corresponding miRNA expression. Our framework was further validated with functional analysis and augmented with rich features including the ability to analyze miRNA isoforms; aggregative prognostic analysis; advanced annotations such as mutation type, regulator alteration, somatic motif, and disease association; and instructive visualization such as mutation OncoPrint, Ideogram, and interactive mRNA-miRNA network.</p><p><strong>Availability and implementation: </strong>The data underlying this article are available in MutMix, at http://innovebioinfo.com/Database/TmiEx/MutMix.php.</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10474951/pdf/","citationCount":"0","resultStr":"{\"title\":\"Somatic mutation effects diffused over microRNA dysregulation.\",\"authors\":\"Hui Yu, Limin Jiang, Chung-I Li, Scott Ness, Sara G M Piccirillo, Yan Guo\",\"doi\":\"10.1093/bioinformatics/btad520\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>As an important player in transcriptome regulation, microRNAs may effectively diffuse somatic mutation impacts to broad cellular processes and ultimately manifest disease and dictate prognosis. Previous studies that tried to correlate mutation with gene expression dysregulation neglected to adjust for the disparate multitudes of false positives associated with unequal sample sizes and uneven class balancing scenarios.</p><p><strong>Results: </strong>To properly address this issue, we developed a statistical framework to rigorously assess the extent of mutation impact on microRNAs in relation to a permutation-based null distribution of a matching sample structure. Carrying out the framework in a pan-cancer study, we ascertained 9008 protein-coding genes with statistically significant mutation impacts on miRNAs. Of these, the collective miRNA expression for 83 genes showed significant prognostic power in nine cancer types. For example, in lower-grade glioma, 10 genes' mutations broadly impacted miRNAs, all of which showed prognostic value with the corresponding miRNA expression. Our framework was further validated with functional analysis and augmented with rich features including the ability to analyze miRNA isoforms; aggregative prognostic analysis; advanced annotations such as mutation type, regulator alteration, somatic motif, and disease association; and instructive visualization such as mutation OncoPrint, Ideogram, and interactive mRNA-miRNA network.</p><p><strong>Availability and implementation: </strong>The data underlying this article are available in MutMix, at http://innovebioinfo.com/Database/TmiEx/MutMix.php.</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 9\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10474951/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad520\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad520","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Somatic mutation effects diffused over microRNA dysregulation.
Motivation: As an important player in transcriptome regulation, microRNAs may effectively diffuse somatic mutation impacts to broad cellular processes and ultimately manifest disease and dictate prognosis. Previous studies that tried to correlate mutation with gene expression dysregulation neglected to adjust for the disparate multitudes of false positives associated with unequal sample sizes and uneven class balancing scenarios.
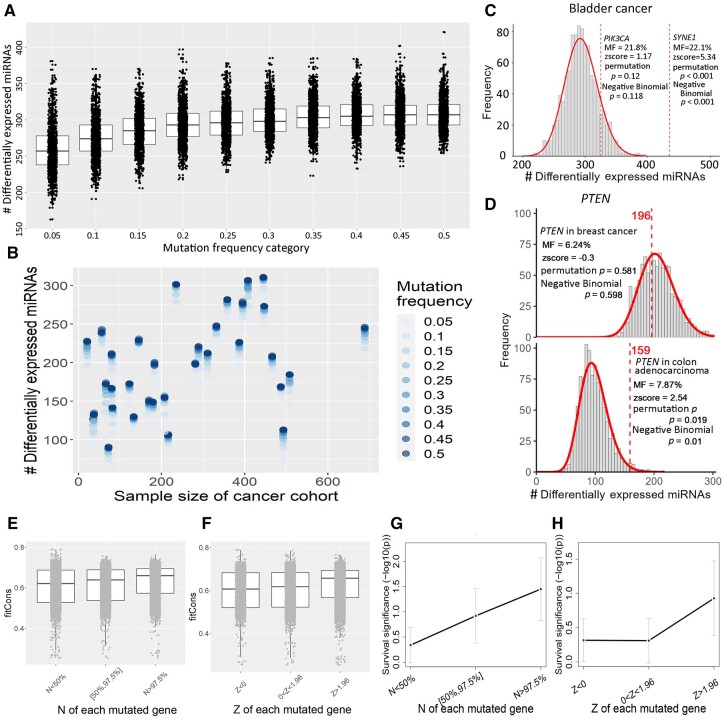
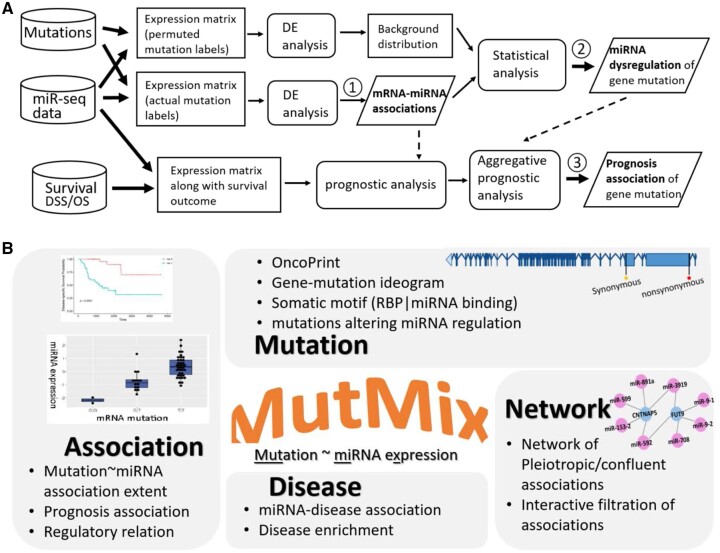
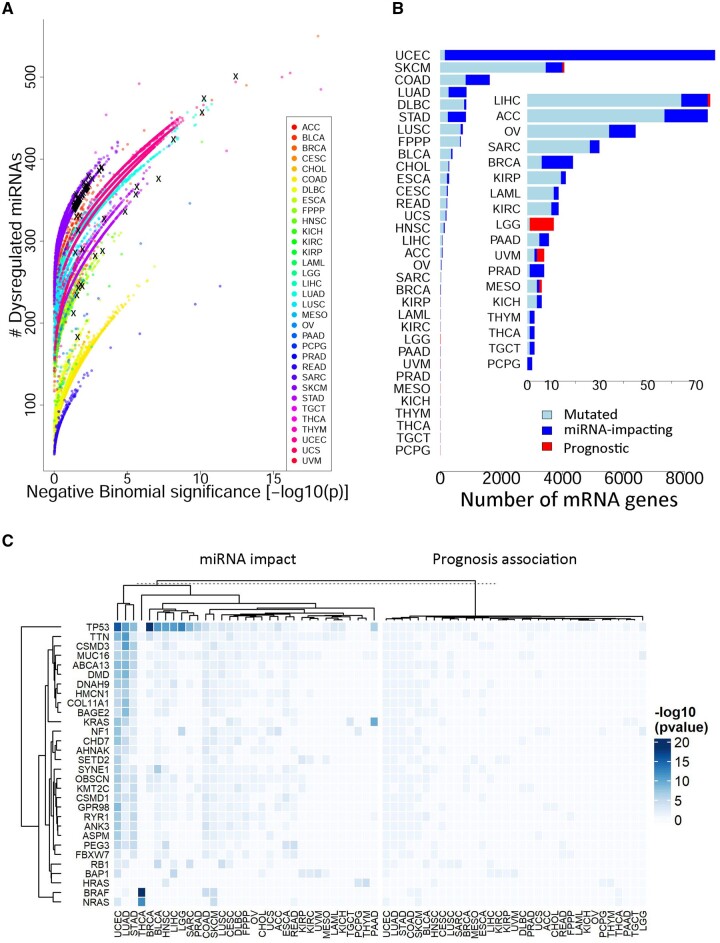
Results: To properly address this issue, we developed a statistical framework to rigorously assess the extent of mutation impact on microRNAs in relation to a permutation-based null distribution of a matching sample structure. Carrying out the framework in a pan-cancer study, we ascertained 9008 protein-coding genes with statistically significant mutation impacts on miRNAs. Of these, the collective miRNA expression for 83 genes showed significant prognostic power in nine cancer types. For example, in lower-grade glioma, 10 genes' mutations broadly impacted miRNAs, all of which showed prognostic value with the corresponding miRNA expression. Our framework was further validated with functional analysis and augmented with rich features including the ability to analyze miRNA isoforms; aggregative prognostic analysis; advanced annotations such as mutation type, regulator alteration, somatic motif, and disease association; and instructive visualization such as mutation OncoPrint, Ideogram, and interactive mRNA-miRNA network.
Availability and implementation: The data underlying this article are available in MutMix, at http://innovebioinfo.com/Database/TmiEx/MutMix.php.
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


