Sara Jones, Matthew Beyers, Maulik Shukla, Fangfang Xia, Thomas Brettin, Rick Stevens, M Ryan Weil, Satishkumar Ranganathan Ganakammal
{"title":"TULIP:使用卷积神经网络的基于rna序列的原发性肿瘤类型预测工具。","authors":"Sara Jones, Matthew Beyers, Maulik Shukla, Fangfang Xia, Thomas Brettin, Rick Stevens, M Ryan Weil, Satishkumar Ranganathan Ganakammal","doi":"10.1177/11769351221139491","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>With cancer as one of the leading causes of death worldwide, accurate primary tumor type prediction is critical in identifying genetic factors that can inhibit or slow tumor progression. There have been efforts to categorize primary tumor types with gene expression data using machine learning, and more recently with deep learning, in the last several years.</p><p><strong>Methods: </strong>In this paper, we developed four 1-dimensional (1D) Convolutional Neural Network (CNN) models to classify RNA-seq count data as one of 17 highly represented primary tumor types or 32 primary tumor types regardless of imbalanced representation. Additionally, we adapted the models to take as input either all Ensembl genes (60,483) or protein coding genes only (19,758). Unlike previous work, we avoided selection bias by not filtering genes based on expression values. RNA-seq count data expressed as FPKM-UQ of 9,025 and 10,940 samples from The Cancer Genome Atlas (TCGA) were downloaded from the Genomic Data Commons (GDC) corresponding to 17 and 32 primary tumor types respectively for training and validating the models.</p><p><strong>Results: </strong>All 4 1D-CNN models had an overall accuracy of 94.7% to 97.6% on the test dataset. Further evaluation indicates that the models with protein coding genes only as features performed with better accuracy compared to the models with all Ensembl genes for both 17 and 32 primary tumor types. For all models, the accuracy by primary tumor type was above 80% for most primary tumor types.</p><p><strong>Conclusions: </strong>We packaged all 4 models as a Python-based deep learning classification tool called TULIP (TUmor CLassIfication Predictor) for performing quality control on primary tumor samples and characterizing cancer samples of unknown tumor type. Further optimization of the models is needed to improve the accuracy of certain primary tumor types.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"21 ","pages":"11769351221139491"},"PeriodicalIF":2.5000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/0b/10.1177_11769351221139491.PMC9729992.pdf","citationCount":"2","resultStr":"{\"title\":\"TULIP: An RNA-seq-based Primary Tumor Type Prediction Tool Using Convolutional Neural Networks.\",\"authors\":\"Sara Jones, Matthew Beyers, Maulik Shukla, Fangfang Xia, Thomas Brettin, Rick Stevens, M Ryan Weil, Satishkumar Ranganathan Ganakammal\",\"doi\":\"10.1177/11769351221139491\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>With cancer as one of the leading causes of death worldwide, accurate primary tumor type prediction is critical in identifying genetic factors that can inhibit or slow tumor progression. There have been efforts to categorize primary tumor types with gene expression data using machine learning, and more recently with deep learning, in the last several years.</p><p><strong>Methods: </strong>In this paper, we developed four 1-dimensional (1D) Convolutional Neural Network (CNN) models to classify RNA-seq count data as one of 17 highly represented primary tumor types or 32 primary tumor types regardless of imbalanced representation. Additionally, we adapted the models to take as input either all Ensembl genes (60,483) or protein coding genes only (19,758). Unlike previous work, we avoided selection bias by not filtering genes based on expression values. RNA-seq count data expressed as FPKM-UQ of 9,025 and 10,940 samples from The Cancer Genome Atlas (TCGA) were downloaded from the Genomic Data Commons (GDC) corresponding to 17 and 32 primary tumor types respectively for training and validating the models.</p><p><strong>Results: </strong>All 4 1D-CNN models had an overall accuracy of 94.7% to 97.6% on the test dataset. Further evaluation indicates that the models with protein coding genes only as features performed with better accuracy compared to the models with all Ensembl genes for both 17 and 32 primary tumor types. For all models, the accuracy by primary tumor type was above 80% for most primary tumor types.</p><p><strong>Conclusions: </strong>We packaged all 4 models as a Python-based deep learning classification tool called TULIP (TUmor CLassIfication Predictor) for performing quality control on primary tumor samples and characterizing cancer samples of unknown tumor type. Further optimization of the models is needed to improve the accuracy of certain primary tumor types.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"21 \",\"pages\":\"11769351221139491\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/0b/10.1177_11769351221139491.PMC9729992.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351221139491\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351221139491","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
TULIP: An RNA-seq-based Primary Tumor Type Prediction Tool Using Convolutional Neural Networks.
Background: With cancer as one of the leading causes of death worldwide, accurate primary tumor type prediction is critical in identifying genetic factors that can inhibit or slow tumor progression. There have been efforts to categorize primary tumor types with gene expression data using machine learning, and more recently with deep learning, in the last several years.
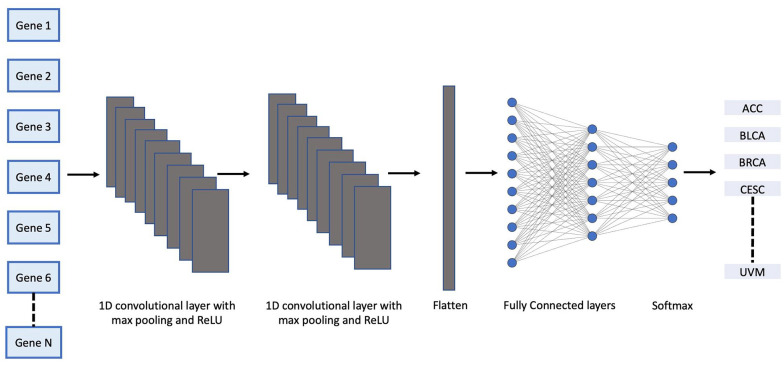
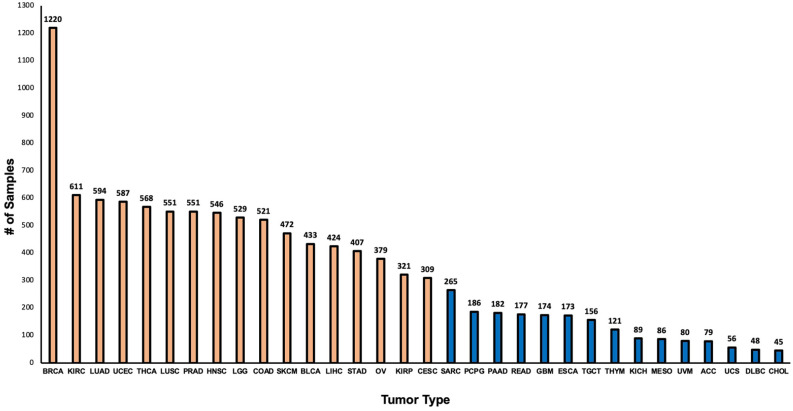
Methods: In this paper, we developed four 1-dimensional (1D) Convolutional Neural Network (CNN) models to classify RNA-seq count data as one of 17 highly represented primary tumor types or 32 primary tumor types regardless of imbalanced representation. Additionally, we adapted the models to take as input either all Ensembl genes (60,483) or protein coding genes only (19,758). Unlike previous work, we avoided selection bias by not filtering genes based on expression values. RNA-seq count data expressed as FPKM-UQ of 9,025 and 10,940 samples from The Cancer Genome Atlas (TCGA) were downloaded from the Genomic Data Commons (GDC) corresponding to 17 and 32 primary tumor types respectively for training and validating the models.
Results: All 4 1D-CNN models had an overall accuracy of 94.7% to 97.6% on the test dataset. Further evaluation indicates that the models with protein coding genes only as features performed with better accuracy compared to the models with all Ensembl genes for both 17 and 32 primary tumor types. For all models, the accuracy by primary tumor type was above 80% for most primary tumor types.
Conclusions: We packaged all 4 models as a Python-based deep learning classification tool called TULIP (TUmor CLassIfication Predictor) for performing quality control on primary tumor samples and characterizing cancer samples of unknown tumor type. Further optimization of the models is needed to improve the accuracy of certain primary tumor types.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


