{"title":"阿霉素诱导的转录组与相互作用组:新药物靶点的鉴定。","authors":"Hilal Taymaz-Nikerel","doi":"10.3906/biy-2107-45","DOIUrl":null,"url":null,"abstract":"<p><p>The working mechanism of the chemotherapeutic drug doxorubicin, which is frequently used in cancer treatment, its effects on cell metabolism, and pathways activated solely by doxorubicin are not fully known. Understanding these principles is important both in improving existing therapies and in finding new drug targets. Here, I describe a systems-biology approach to find a generalizable working principle for doxorubicin by superimposition of human interactome over gene datasets commonly expressed among various cancer types. The common -in at least two different diseases-transcriptional response of distinctive cancer cell lines to doxorubicin was reflected via 199 significantly and differentially expressed genes, mostly related to the regulation of transcription. Then, by integrating with interactome data, an active network was constructed allowing detection of clusters. Since each cluster defines densely connected regions, another level of understanding of functional principles is provided. Significant clusters were associated with the linked transcription factors and transcriptional factor enrichment analysis within these regulatory networks led to the proposition of Pou5f1b, Znf428, Prmt3, Znf12, Erg, Tfdp1, Foxm1, and Cenpa as new drug targets in drug development that can be applied in different cancer types.</p>","PeriodicalId":23375,"journal":{"name":"Turkish journal of biology = Turk biyoloji dergisi","volume":"46 2","pages":"137-144"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10393105/pdf/","citationCount":"0","resultStr":"{\"title\":\"Doxorubicin-induced transcriptome meets interactome: identification of new drug targets.\",\"authors\":\"Hilal Taymaz-Nikerel\",\"doi\":\"10.3906/biy-2107-45\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The working mechanism of the chemotherapeutic drug doxorubicin, which is frequently used in cancer treatment, its effects on cell metabolism, and pathways activated solely by doxorubicin are not fully known. Understanding these principles is important both in improving existing therapies and in finding new drug targets. Here, I describe a systems-biology approach to find a generalizable working principle for doxorubicin by superimposition of human interactome over gene datasets commonly expressed among various cancer types. The common -in at least two different diseases-transcriptional response of distinctive cancer cell lines to doxorubicin was reflected via 199 significantly and differentially expressed genes, mostly related to the regulation of transcription. Then, by integrating with interactome data, an active network was constructed allowing detection of clusters. Since each cluster defines densely connected regions, another level of understanding of functional principles is provided. Significant clusters were associated with the linked transcription factors and transcriptional factor enrichment analysis within these regulatory networks led to the proposition of Pou5f1b, Znf428, Prmt3, Znf12, Erg, Tfdp1, Foxm1, and Cenpa as new drug targets in drug development that can be applied in different cancer types.</p>\",\"PeriodicalId\":23375,\"journal\":{\"name\":\"Turkish journal of biology = Turk biyoloji dergisi\",\"volume\":\"46 2\",\"pages\":\"137-144\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10393105/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Turkish journal of biology = Turk biyoloji dergisi\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3906/biy-2107-45\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Turkish journal of biology = Turk biyoloji dergisi","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3906/biy-2107-45","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Doxorubicin-induced transcriptome meets interactome: identification of new drug targets.
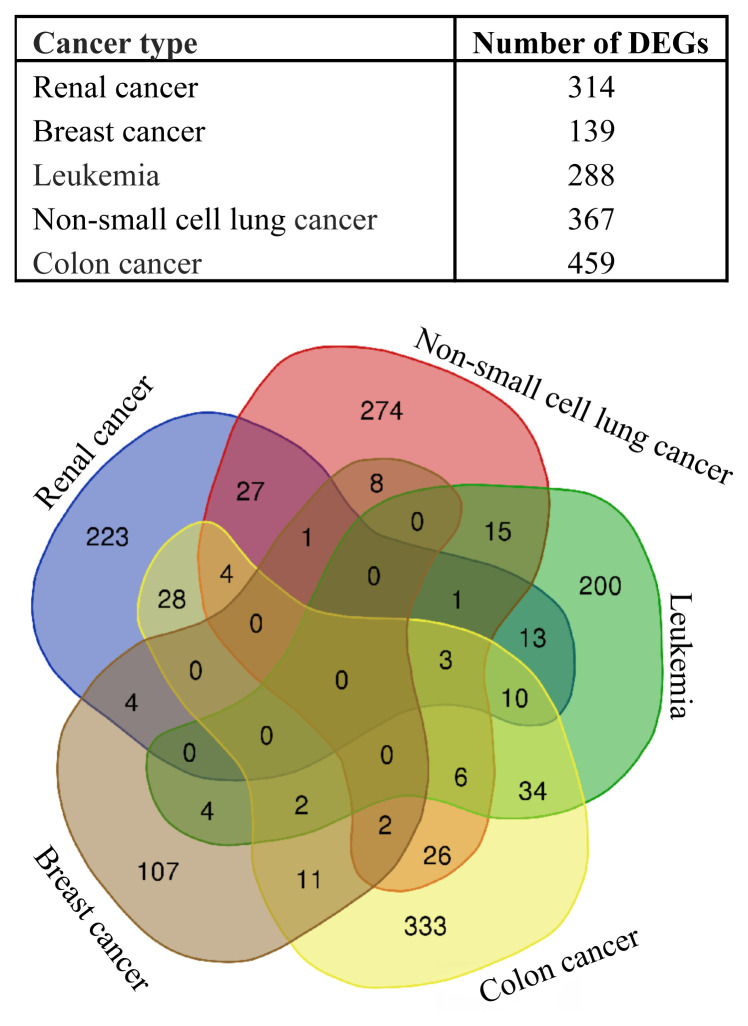
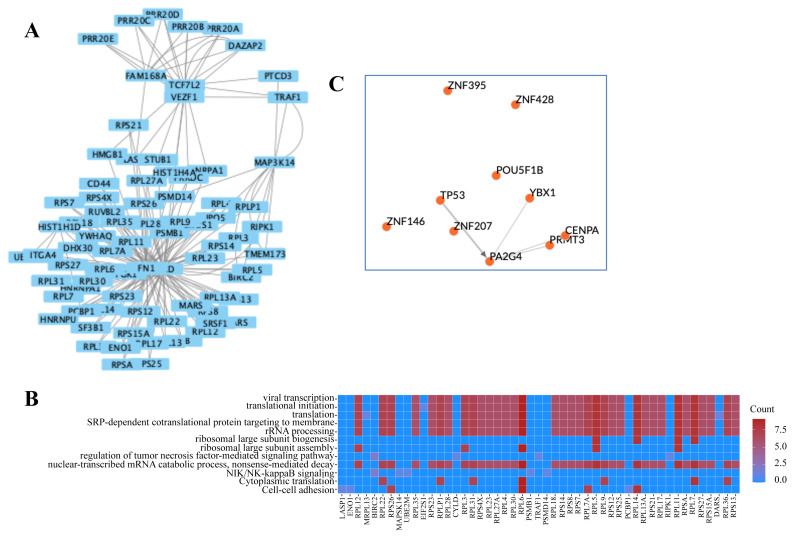
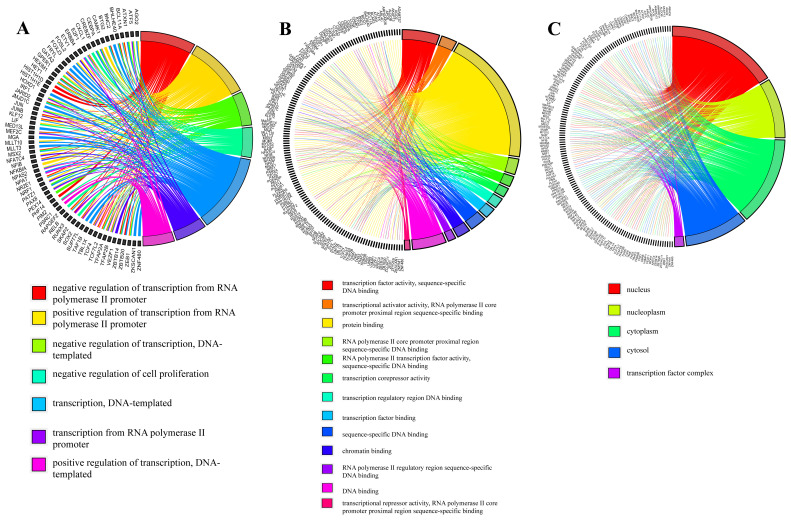
The working mechanism of the chemotherapeutic drug doxorubicin, which is frequently used in cancer treatment, its effects on cell metabolism, and pathways activated solely by doxorubicin are not fully known. Understanding these principles is important both in improving existing therapies and in finding new drug targets. Here, I describe a systems-biology approach to find a generalizable working principle for doxorubicin by superimposition of human interactome over gene datasets commonly expressed among various cancer types. The common -in at least two different diseases-transcriptional response of distinctive cancer cell lines to doxorubicin was reflected via 199 significantly and differentially expressed genes, mostly related to the regulation of transcription. Then, by integrating with interactome data, an active network was constructed allowing detection of clusters. Since each cluster defines densely connected regions, another level of understanding of functional principles is provided. Significant clusters were associated with the linked transcription factors and transcriptional factor enrichment analysis within these regulatory networks led to the proposition of Pou5f1b, Znf428, Prmt3, Znf12, Erg, Tfdp1, Foxm1, and Cenpa as new drug targets in drug development that can be applied in different cancer types.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


