群体人口学和适应性推断的全基因组和减少代表性测序对比:一项高山哺乳动物案例研究。
IF 3.9
2区 生物学
Q2 ECOLOGY
引用次数: 2
摘要
基因组记录了一个物种的适应性和人口统计学历史,但测序策略和样本量的选择可能会影响这种推断。我们比较了全基因组和减少代表性测序方法,以研究北美山山羊(Oreamnos americanus)的种群人口统计和适应性信号。我们对254个个体应用了限制性位点相关DNA测序(RADseq)方法,对35个物种范围内的中等覆盖率个体应用了全基因组重测序(WGS)方法(9X),对5个高覆盖率个体(30X)。我们使用ANGSD来估计基因型可能性,估计有效种群规模(Ne)、种群结构,并用δaδi和MSMC2明确建模人口统计学历史。数据集在支持冰川引起的替代作用和山羊极低Ne方面总体一致。我们使用冗余分析评估了一组气候变量和地理位置作为遗传多样性的预测因素。地理和气候变量解释了总方差的中等比例(WGS为36%,RADseq数据集为21%);这两个数据集都支持漂移的巨大影响和一定程度的局部适应。本文提出的WGS和RADseq的经验相似性令人放心地表明,这两种方法都将在人群中恢复大量的人口统计和自适应信号;然而,与RADseq相比,WGS提供了一些优势,例如推断自适应过程和计算纯合性估计的运行次数。考虑到预测的气候导致的高山环境变化和遗传上不成熟的山羊,这个神秘物种的长期适应能力值得怀疑。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Contrasting whole-genome and reduced representation sequencing for population demographic and adaptive inference: an alpine mammal case study
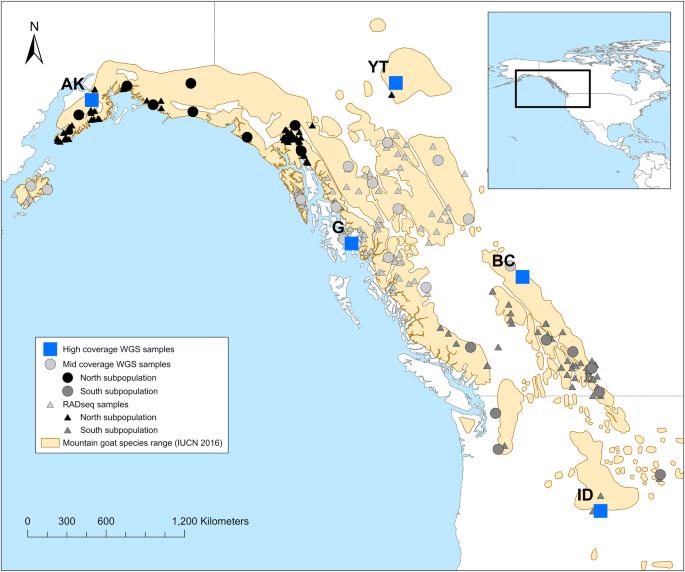
Genomes capture the adaptive and demographic history of a species, but the choice of sequencing strategy and sample size can impact such inferences. We compared whole genome and reduced representation sequencing approaches to study the population demographic and adaptive signals of the North American mountain goat (Oreamnos americanus). We applied the restriction site-associated DNA sequencing (RADseq) approach to 254 individuals and whole genome resequencing (WGS) approach to 35 individuals across the species range at mid-level coverage (9X) and to 5 individuals at high coverage (30X). We used ANGSD to estimate the genotype likelihoods and estimated the effective population size (Ne), population structure, and explicitly modelled the demographic history with δaδi and MSMC2. The data sets were overall concordant in supporting a glacial induced vicariance and extremely low Ne in mountain goats. We evaluated a set of climatic variables and geographic location as predictors of genetic diversity using redundancy analysis. A moderate proportion of total variance (36% for WGS and 21% for RADseq data sets) was explained by geography and climate variables; both data sets support a large impact of drift and some degree of local adaptation. The empirical similarities of WGS and RADseq presented herein reassuringly suggest that both approaches will recover large demographic and adaptive signals in a population; however, WGS offers several advantages over RADseq, such as inferring adaptive processes and calculating runs-of-homozygosity estimates. Considering the predicted climate-induced changes in alpine environments and the genetically depauperate mountain goat, the long-term adaptive capabilities of this enigmatic species are questionable.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Heredity
生物-进化生物学
CiteScore
7.50
自引率
2.60%
发文量
84
审稿时长
4-8 weeks
期刊介绍:
Heredity is the official journal of the Genetics Society. It covers a broad range of topics within the field of genetics and therefore papers must address conceptual or applied issues of interest to the journal''s wide readership
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


