Yan Shen, Il-Man Kim, Neal L Weintraub, Yaoliang Tang
{"title":"通过空间单细胞转录组学鉴定人类梗死心脏中存活心肌细胞的代谢状态。","authors":"Yan Shen, Il-Man Kim, Neal L Weintraub, Yaoliang Tang","doi":"10.1097/CP9.0000000000000038","DOIUrl":null,"url":null,"abstract":"<p><p>The metabolic status of surviving cardiomyocytes (CM) in the myocardial tissues of patients who sustained myocardial infarction (MI) is largely unknown. Spatial single-cell RNA-sequencing (scRNA-seq) is a novel tool that enables the unbiased analysis of RNA signatures within intact tissues. We employed this tool to assess the metabolic profiles of surviving CM in the myocardial tissues of patients post-MI.</p><p><strong>Methods: </strong>A spatial scRNA-seq dataset was used to compare the genetic profiles of CM from patients with MI and control patients; we analyzed the metabolic adaptations of surviving CM within the ischemic niche. A standard pipeline in Seurat was used for data analysis, including normalization, feature selection, and identification of highly variable genes using principal component analysis (PCA). Harmony was used to remove batch effects and integrate the CM samples based on annotations. Uniform manifold approximation and projection (UMAP) was used for dimensional reduction. The Seurat \"FindMarkers\" function was used to identify differentially expressed genes (DEGs), which were analyzed by the Gene Ontology (GO) enrichment pathway. Finally, the scMetabolism R tool pipeline with parameters method = VISION (Vision is a flexible system that utilizes a high-throughput pipeline and an interactive web-based report to annotate and explore scRNA-seq datasets in a dynamic manner) and metabolism.type = Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to quantify the metabolic activity of each CM.</p><p><strong>Results: </strong>Analysis of spatial scRNA-seq data showed fewer surviving CM in infarcted hearts than in control hearts. GO analysis revealed repressed pathways in oxidative phosphorylation, cardiac cell development, and activated pathways in response to stimuli and macromolecular metabolic processes. Metabolic analysis showed downregulated energy and amino acid pathways and increased purine, pyrimidine, and one-carbon pool by folate pathways in surviving CM.</p><p><strong>Conclusions: </strong>Surviving CM within the infarcted myocardium exhibited metabolic adaptations, as evidenced by the downregulation of most pathways linked to oxidative phosphorylation, glucose, fatty acid, and amino acid metabolism. In contrast, pathways linked to purine and pyrimidine metabolism, fatty acid biosynthesis, and one-carbon metabolism were upregulated in surviving CM. These novel findings have implications for the development of effective strategies to improve the survival of hibernating CM within the infarcted heart.</p>","PeriodicalId":52908,"journal":{"name":"Cardiology Plus","volume":"8 1","pages":"18-26"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7d/ed/cp9-8-18.PMC10180026.pdf","citationCount":"2","resultStr":"{\"title\":\"Identification of the metabolic state of surviving cardiomyocytes in the human infarcted heart by spatial single-cell transcriptomics.\",\"authors\":\"Yan Shen, Il-Man Kim, Neal L Weintraub, Yaoliang Tang\",\"doi\":\"10.1097/CP9.0000000000000038\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The metabolic status of surviving cardiomyocytes (CM) in the myocardial tissues of patients who sustained myocardial infarction (MI) is largely unknown. Spatial single-cell RNA-sequencing (scRNA-seq) is a novel tool that enables the unbiased analysis of RNA signatures within intact tissues. We employed this tool to assess the metabolic profiles of surviving CM in the myocardial tissues of patients post-MI.</p><p><strong>Methods: </strong>A spatial scRNA-seq dataset was used to compare the genetic profiles of CM from patients with MI and control patients; we analyzed the metabolic adaptations of surviving CM within the ischemic niche. A standard pipeline in Seurat was used for data analysis, including normalization, feature selection, and identification of highly variable genes using principal component analysis (PCA). Harmony was used to remove batch effects and integrate the CM samples based on annotations. Uniform manifold approximation and projection (UMAP) was used for dimensional reduction. The Seurat \\\"FindMarkers\\\" function was used to identify differentially expressed genes (DEGs), which were analyzed by the Gene Ontology (GO) enrichment pathway. Finally, the scMetabolism R tool pipeline with parameters method = VISION (Vision is a flexible system that utilizes a high-throughput pipeline and an interactive web-based report to annotate and explore scRNA-seq datasets in a dynamic manner) and metabolism.type = Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to quantify the metabolic activity of each CM.</p><p><strong>Results: </strong>Analysis of spatial scRNA-seq data showed fewer surviving CM in infarcted hearts than in control hearts. GO analysis revealed repressed pathways in oxidative phosphorylation, cardiac cell development, and activated pathways in response to stimuli and macromolecular metabolic processes. Metabolic analysis showed downregulated energy and amino acid pathways and increased purine, pyrimidine, and one-carbon pool by folate pathways in surviving CM.</p><p><strong>Conclusions: </strong>Surviving CM within the infarcted myocardium exhibited metabolic adaptations, as evidenced by the downregulation of most pathways linked to oxidative phosphorylation, glucose, fatty acid, and amino acid metabolism. In contrast, pathways linked to purine and pyrimidine metabolism, fatty acid biosynthesis, and one-carbon metabolism were upregulated in surviving CM. These novel findings have implications for the development of effective strategies to improve the survival of hibernating CM within the infarcted heart.</p>\",\"PeriodicalId\":52908,\"journal\":{\"name\":\"Cardiology Plus\",\"volume\":\"8 1\",\"pages\":\"18-26\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/7d/ed/cp9-8-18.PMC10180026.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cardiology Plus\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1097/CP9.0000000000000038\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/4/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cardiology Plus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1097/CP9.0000000000000038","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/4/4 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 2
摘要
心肌梗死(MI)患者心肌组织中存活心肌细胞(CM)的代谢状况在很大程度上是未知的。空间单细胞RNA测序(scRNA-seq)是一种新的工具,能够对完整组织内的RNA特征进行无偏分析。我们使用该工具评估MI后患者心肌组织中存活CM的代谢谱。方法:使用空间scRNA-seq数据集比较MI患者和对照患者CM的遗传谱;我们分析了存活的CM在缺血生态位内的代谢适应。Seurat中的标准管道用于数据分析,包括归一化、特征选择和使用主成分分析(PCA)识别高度可变基因。Harmony用于消除批量效应,并基于注释集成CM样本。统一流形近似和投影(UMAP)用于降维。Seurat“FindMarkers”功能用于鉴定差异表达基因(DEG),通过基因本体论(GO)富集途径对其进行分析。最后scMetabolism R工具管道的参数方法=VISION(VISION是一个灵活的系统,它利用高通量管道和交互式网络报告以动态方式注释和探索scRNA-seq数据集)和metabolism.type=Kyoto Encyclopedia of Genes and Genomes(KEGG)来量化每个CM的代谢活性数据显示梗死心脏中存活的CM少于对照心脏。GO分析揭示了氧化磷酸化、心脏细胞发育中被抑制的途径,以及刺激和大分子代谢过程中被激活的途径。代谢分析显示,在存活的CM中,能量和氨基酸途径下调,嘌呤、嘧啶和一个碳库被叶酸途径增加。结论:梗死心肌中存活的CM表现出代谢适应,与氧化磷酸化、葡萄糖、脂肪酸和氨基酸代谢相关的大多数途径下调就是明证。相反,在存活的CM中,与嘌呤和嘧啶代谢、脂肪酸生物合成和单碳代谢相关的途径被上调。这些新发现对开发有效策略以提高梗死心脏内冬眠CM的存活率具有启示意义。
Identification of the metabolic state of surviving cardiomyocytes in the human infarcted heart by spatial single-cell transcriptomics.
The metabolic status of surviving cardiomyocytes (CM) in the myocardial tissues of patients who sustained myocardial infarction (MI) is largely unknown. Spatial single-cell RNA-sequencing (scRNA-seq) is a novel tool that enables the unbiased analysis of RNA signatures within intact tissues. We employed this tool to assess the metabolic profiles of surviving CM in the myocardial tissues of patients post-MI.
Methods: A spatial scRNA-seq dataset was used to compare the genetic profiles of CM from patients with MI and control patients; we analyzed the metabolic adaptations of surviving CM within the ischemic niche. A standard pipeline in Seurat was used for data analysis, including normalization, feature selection, and identification of highly variable genes using principal component analysis (PCA). Harmony was used to remove batch effects and integrate the CM samples based on annotations. Uniform manifold approximation and projection (UMAP) was used for dimensional reduction. The Seurat "FindMarkers" function was used to identify differentially expressed genes (DEGs), which were analyzed by the Gene Ontology (GO) enrichment pathway. Finally, the scMetabolism R tool pipeline with parameters method = VISION (Vision is a flexible system that utilizes a high-throughput pipeline and an interactive web-based report to annotate and explore scRNA-seq datasets in a dynamic manner) and metabolism.type = Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to quantify the metabolic activity of each CM.
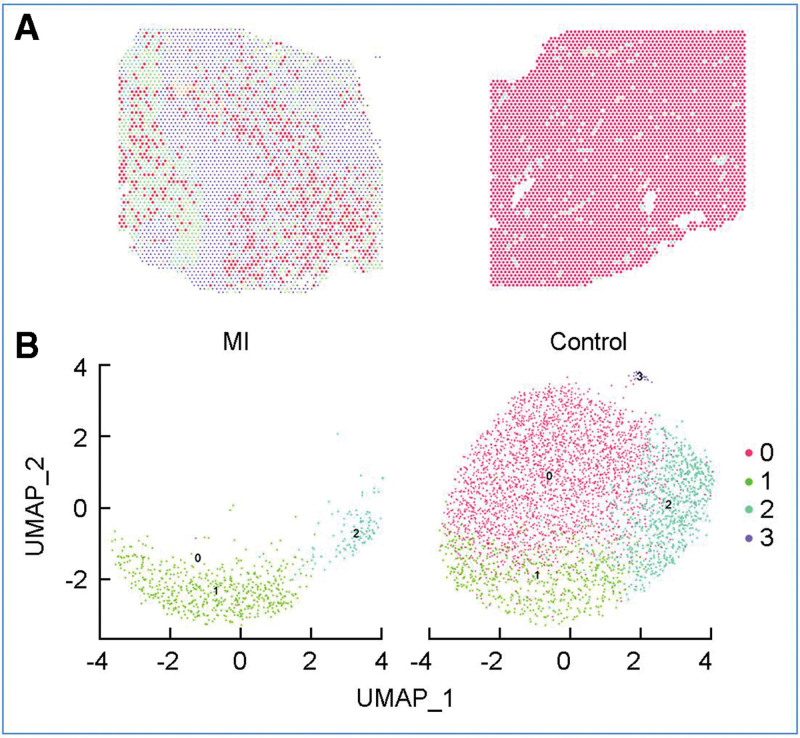
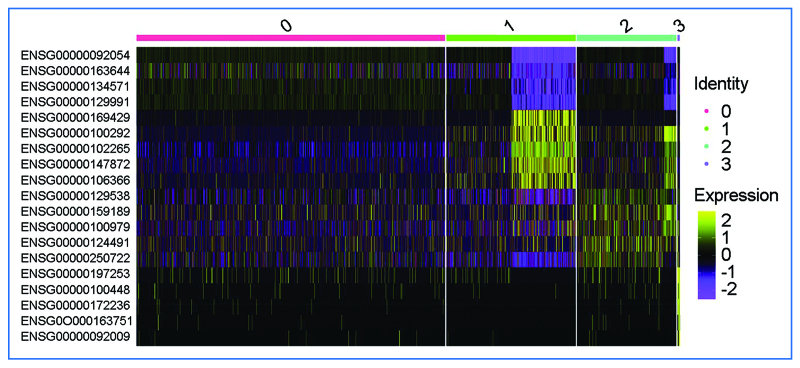
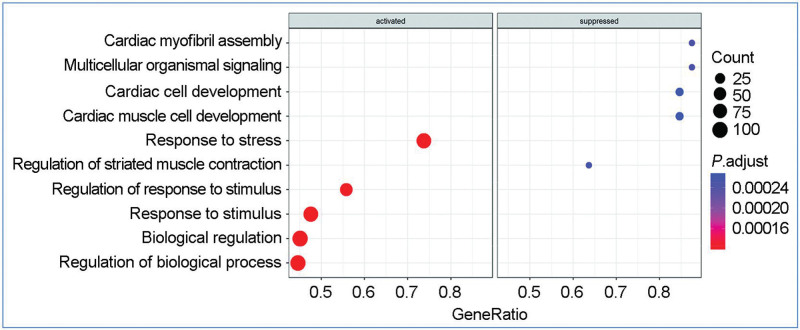
Results: Analysis of spatial scRNA-seq data showed fewer surviving CM in infarcted hearts than in control hearts. GO analysis revealed repressed pathways in oxidative phosphorylation, cardiac cell development, and activated pathways in response to stimuli and macromolecular metabolic processes. Metabolic analysis showed downregulated energy and amino acid pathways and increased purine, pyrimidine, and one-carbon pool by folate pathways in surviving CM.
Conclusions: Surviving CM within the infarcted myocardium exhibited metabolic adaptations, as evidenced by the downregulation of most pathways linked to oxidative phosphorylation, glucose, fatty acid, and amino acid metabolism. In contrast, pathways linked to purine and pyrimidine metabolism, fatty acid biosynthesis, and one-carbon metabolism were upregulated in surviving CM. These novel findings have implications for the development of effective strategies to improve the survival of hibernating CM within the infarcted heart.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


