Weiwen Wang, James Barbetti, Thomas Wong, Bryan Thornlow, Russ Corbett-Detig, Yatish Turakhia, Robert Lanfear, Bui Quang Minh
{"title":"DecentTree:基因组时代可扩展的邻居加入。","authors":"Weiwen Wang, James Barbetti, Thomas Wong, Bryan Thornlow, Russ Corbett-Detig, Yatish Turakhia, Robert Lanfear, Bui Quang Minh","doi":"10.1093/bioinformatics/btad536","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Neighbour-Joining is one of the most widely used distance-based phylogenetic inference methods. However, current implementations do not scale well for datasets with more than 10 000 sequences. Given the increasing pace of generating new sequence data, particularly in outbreaks of emerging diseases, and the already enormous existing databases of sequence data for which Neighbour-Joining is a useful approach, new implementations of existing methods are warranted.</p><p><strong>Results: </strong>Here, we present DecentTree, which provides highly optimized and parallel implementations of Neighbour-Joining and several of its variants. DecentTree is designed as a stand-alone application and a header-only library easily integrated with other phylogenetic software (e.g. it is integral in the popular IQ-TREE software). We show that DecentTree shows similar or improved performance over existing software (BIONJ, Quicktree, FastME, and RapidNJ), especially for handling very large alignments. For example, DecentTree is up to 6-fold faster than the fastest existing Neighbour-Joining software (e.g. RapidNJ) when generating a tree of 64 000 SARS-CoV-2 genomes.</p><p><strong>Availability and implementation: </strong>DecentTree is open source and freely available at https://github.com/iqtree/decenttree. All code and data used in this analysis are available on Github (https://github.com/asdcid/Comparison-of-neighbour-joining-software).</p>","PeriodicalId":8903,"journal":{"name":"Bioinformatics","volume":"39 9","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2023-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491953/pdf/","citationCount":"3","resultStr":"{\"title\":\"DecentTree: scalable Neighbour-Joining for the genomic era.\",\"authors\":\"Weiwen Wang, James Barbetti, Thomas Wong, Bryan Thornlow, Russ Corbett-Detig, Yatish Turakhia, Robert Lanfear, Bui Quang Minh\",\"doi\":\"10.1093/bioinformatics/btad536\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Neighbour-Joining is one of the most widely used distance-based phylogenetic inference methods. However, current implementations do not scale well for datasets with more than 10 000 sequences. Given the increasing pace of generating new sequence data, particularly in outbreaks of emerging diseases, and the already enormous existing databases of sequence data for which Neighbour-Joining is a useful approach, new implementations of existing methods are warranted.</p><p><strong>Results: </strong>Here, we present DecentTree, which provides highly optimized and parallel implementations of Neighbour-Joining and several of its variants. DecentTree is designed as a stand-alone application and a header-only library easily integrated with other phylogenetic software (e.g. it is integral in the popular IQ-TREE software). We show that DecentTree shows similar or improved performance over existing software (BIONJ, Quicktree, FastME, and RapidNJ), especially for handling very large alignments. For example, DecentTree is up to 6-fold faster than the fastest existing Neighbour-Joining software (e.g. RapidNJ) when generating a tree of 64 000 SARS-CoV-2 genomes.</p><p><strong>Availability and implementation: </strong>DecentTree is open source and freely available at https://github.com/iqtree/decenttree. All code and data used in this analysis are available on Github (https://github.com/asdcid/Comparison-of-neighbour-joining-software).</p>\",\"PeriodicalId\":8903,\"journal\":{\"name\":\"Bioinformatics\",\"volume\":\"39 9\",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2023-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491953/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btad536\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btad536","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
DecentTree: scalable Neighbour-Joining for the genomic era.
Motivation: Neighbour-Joining is one of the most widely used distance-based phylogenetic inference methods. However, current implementations do not scale well for datasets with more than 10 000 sequences. Given the increasing pace of generating new sequence data, particularly in outbreaks of emerging diseases, and the already enormous existing databases of sequence data for which Neighbour-Joining is a useful approach, new implementations of existing methods are warranted.
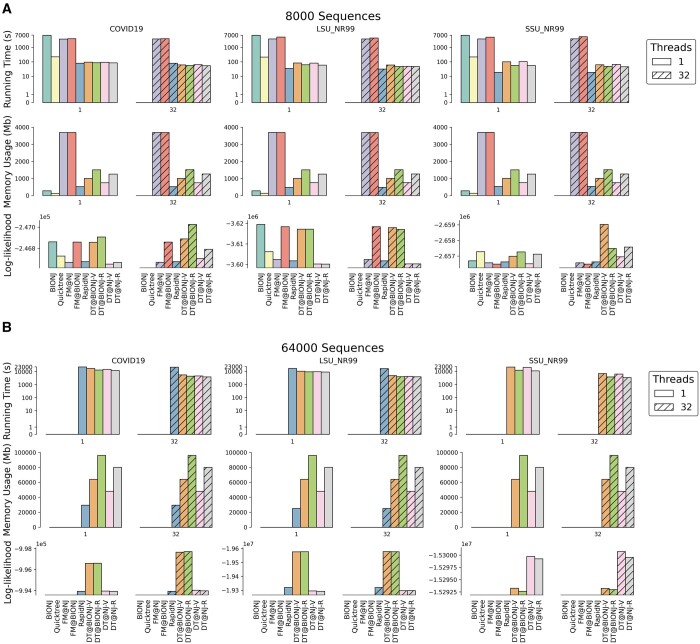
Results: Here, we present DecentTree, which provides highly optimized and parallel implementations of Neighbour-Joining and several of its variants. DecentTree is designed as a stand-alone application and a header-only library easily integrated with other phylogenetic software (e.g. it is integral in the popular IQ-TREE software). We show that DecentTree shows similar or improved performance over existing software (BIONJ, Quicktree, FastME, and RapidNJ), especially for handling very large alignments. For example, DecentTree is up to 6-fold faster than the fastest existing Neighbour-Joining software (e.g. RapidNJ) when generating a tree of 64 000 SARS-CoV-2 genomes.
Availability and implementation: DecentTree is open source and freely available at https://github.com/iqtree/decenttree. All code and data used in this analysis are available on Github (https://github.com/asdcid/Comparison-of-neighbour-joining-software).
期刊介绍:
The leading journal in its field, Bioinformatics publishes the highest quality scientific papers and review articles of interest to academic and industrial researchers. Its main focus is on new developments in genome bioinformatics and computational biology. Two distinct sections within the journal - Discovery Notes and Application Notes- focus on shorter papers; the former reporting biologically interesting discoveries using computational methods, the latter exploring the applications used for experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


