Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer
{"title":"CD5 b细胞显性原发性免疫缺陷:MAGT1缺陷谱的一部分。","authors":"Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer","doi":"10.1177/27534030231199675","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the \"XMEN\" title pathologies. The updated title, \"X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect,\" was proposed in 2020.</p><p><strong>Objectives: </strong>To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.</p><p><strong>Methods: </strong>Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.</p><p><strong>Design: </strong>Immune re-evaluation done through flow cytometry and next-generation sequencing.</p><p><strong>Results: </strong>Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.</p><p><strong>Conclusion: </strong>We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.</p>","PeriodicalId":75217,"journal":{"name":"Therapeutic advances in allergy and rhinology","volume":"14 ","pages":"27534030231199675"},"PeriodicalIF":0.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/c5/10.1177_27534030231199675.PMC10496486.pdf","citationCount":"0","resultStr":"{\"title\":\"CD5 B-Cell Predominant Primary Immunodeficiency: Part of the Spectrum of <i>MAGT1</i> Deficiency.\",\"authors\":\"Marija J Rowane, Benjamin C Stewart-Bates, Rayna J Doll, Howard J Meyerson, John S Venglarcik, Meghan Callahan, Lauren Fill, Remie Saab, Hans D Ochs, Robert W Hostoffer\",\"doi\":\"10.1177/27534030231199675\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the \\\"XMEN\\\" title pathologies. The updated title, \\\"X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect,\\\" was proposed in 2020.</p><p><strong>Objectives: </strong>To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.</p><p><strong>Methods: </strong>Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.</p><p><strong>Design: </strong>Immune re-evaluation done through flow cytometry and next-generation sequencing.</p><p><strong>Results: </strong>Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.</p><p><strong>Conclusion: </strong>We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.</p>\",\"PeriodicalId\":75217,\"journal\":{\"name\":\"Therapeutic advances in allergy and rhinology\",\"volume\":\"14 \",\"pages\":\"27534030231199675\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/c5/10.1177_27534030231199675.PMC10496486.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Therapeutic advances in allergy and rhinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/27534030231199675\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"0\",\"JCRName\":\"OTORHINOLARYNGOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Therapeutic advances in allergy and rhinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/27534030231199675","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"0","JCRName":"OTORHINOLARYNGOLOGY","Score":null,"Total":0}
CD5 B-Cell Predominant Primary Immunodeficiency: Part of the Spectrum of MAGT1 Deficiency.
Background: Selective anti-polysaccharide antibody deficiency (SPAD) with CD5 B-cell predominance and autoimmune phenomena was identified in a male cohort first reported by Antall et al in 1999. The phenotypically likewise and genotypically identical X-linked immunodeficiency with magnesium defect, Epstein-Barr Virus infection, and neoplasia (XMEN) disease was defined as a novel primary immunodeficiency (PID) in 2011. Recent studies of the magnesium transporter 1 (MAGT1) gene mutation reveal glycosylation defects contributing to more phenotypic variance than the "XMEN" title pathologies. The updated title, "X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect," was proposed in 2020.
Objectives: To reflect the patient population more accurately, a prospective classification update may consider MAGT1 glycobiological errors contributing to phenotypic variance but also pre-genetic testing era reports with CD5 B-cell predominance.
Methods: Patient 1 from Antall et al presented at 28 years of age for further immunological evaluation of his CD5/CD19 B-cell predominance diagnosed at 5 years old.
Design: Immune re-evaluation done through flow cytometry and next-generation sequencing.
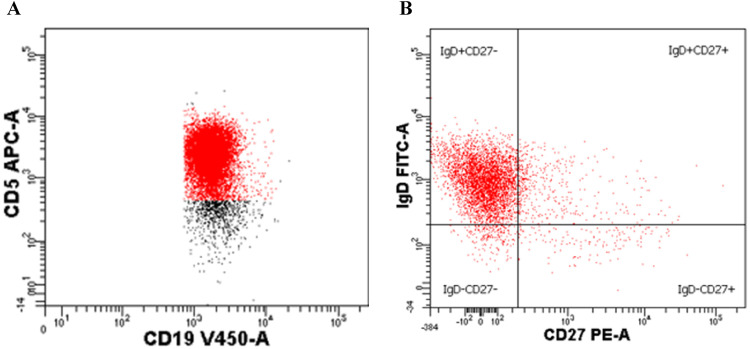
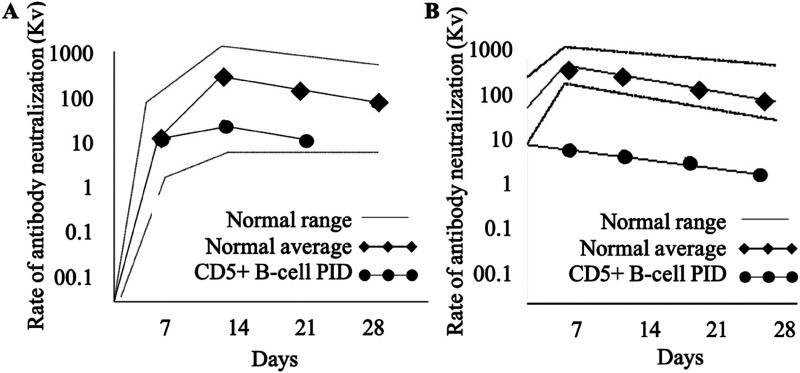
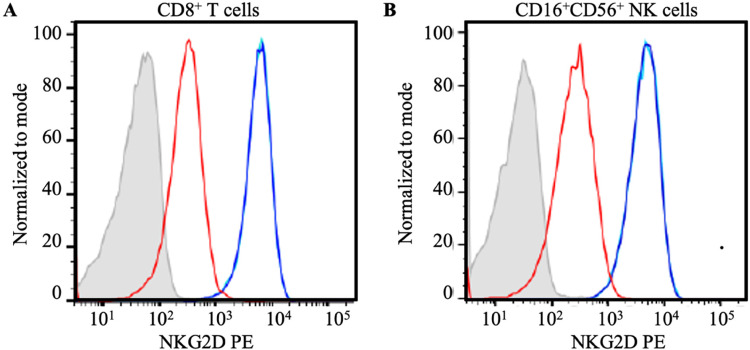
Results: Flow cytometry B-cell phenotyping revealed persistent CD5+CD19+ (93%). Flow cytometric histogram quantified reduced activator CD16+CD56+ natural killer and CD8+ T-cell receptor, Group 2, Member D (NKG2D) glycoprotein expression. A c.923-1_934 deletion loss of function mutation was identified in the MAGT1 gene.
Conclusion: We suggest the novel PID XMEN, based on its CD5 B-cell predominance, had been discovered and reported over a decade earlier as CD5+ PID based on the MAGT1 mutation found in the same. We encourage consideration of combining these labels and recent findings to offer the most accurate classification of this disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


